.png)
Медицинская помощь субъектам клинического испытания

- Библиотека /
-
7258
Украинское законодательство определяет медико-санитарную помощь как комплекс специальных мероприятий, направленных на содействие улучшению здоровья, повышение санитарной культуры, предотвращение заболеваний и инвалидности, на раннюю диагностику, помощь лицам с острыми и хроническими заболеваниями, реабилитацию больных и инвалидов [1]. Поскольку приведенное официальное определение очень обширное, сразу укажем, что в данной статье медицинская помощь субъектам клинического исследования включает прежде всего тщательное обследование испытуемого (в рамках, предусмотренных протоколом исследования, и расширенное обследование при необходимости), постоянный мониторинг состояния здоровья субъектов исследования, своевременную диагностику и лечение побочных явлений, включая серьезные. В случае, если непосредственно лечением развившихся побочных явлений занимаются врачи иной, чем исследователи, специализации, то посильное участие в организации лечения и при необходимости координация усилий «узких» специалистов остаются обязанностью исследователя. Лечение основного заболевания (под основным в данном случае подразумевается заболевание/синдром, которое является показанием к назначению исследуемого препарата) стоит в этом ряду обособлено, так как, с одной стороны, испытуемые добровольно согласились принять участие в клиническом исследовании, то есть получать лечение с недоказанной эффективностью, но, с другой стороны, их здоровье и благополучие максимально гарантируются положениями Хельсинкской декларации (в особенности п. 5, 10 и 19) [2]. Такое кажущееся противоречие между интересами общества (потенциальная польза от результатов исследований с участием человека как субъекта исследования) и превалирующими интересами личности требует от врача-исследователя высокого уровня ответственности и постоянной готовности оказать испытуемым необходимую медицинскую помощь.
Таким образом, предметом обсуждения являются скорее оптимальные формы организации медицинской помощи испытуемым. Анализ положений руководства по надлежащей клинической практике (ICH GCP) также позволит определить круг обязанностей и сферу ответственности спонсора исследования, врача-исследователя и медицинского учреждения в тех случаях, когда испытуемому необходимо оказать медицинскую помощь.
Итак, говоря об оказании медицинской помощи субъектам клинического исследования, необходимо хорошо понимать, что каждый врач-исследователь всегда должен быть готов оказать медицинскую помощь пациенту/здоровому добровольцу в случае необходимости и ни при каких обстоятельствах не может отказать в такой помощи испытуемому.
Важным положением Хельсинкской декларации является п. 17: «Врач должен воздержаться от проведения исследований с участием людей, если он не уверен в том, что риск подобных исследований надежно предсказуем. Врач должен прекратить любые исследования, если обнаруженный риск превышает возможную пользу» [2]. Принимая во внимание тот факт, что клинические испытания практически всегда сопряжены с возможными рисками, так как применяется еще не достаточно изученный и проверенный в клинической практике препарат, одной из наиболее важных для врача-исследователя задач является предвидеть и по возможности предотвращать возможные риски для пациентов, связанные с развитием медицинских осложнений при проведении клинических испытаний. Это подразумевает адекватный подбор пациентов в исследование в соответствии с критериями включения/невключения, тщательное обследование и тщательный сбор анамнеза на этапе отбора и при каждом последующем визите, регулярный анализ результатов проведенных клинико-инструментальных и лабораторных процедур в ходе всего периода исследования. В большинстве случаев тщательное обследование испытуемого предусмотрено протоколом исследования, но при необходимости следует выполнять дополнительные обследования и анализы.
Для проведения клинических исследований в качестве ответственных исследователей привлекаются высококвалифицированные врачи, эксперты в определенной области медицины. Более того, кандидатура каждого исследователя утверждается регуляторным органом и получает одобрение комиссии по вопросам этики.
Проблему оптимальной организации и оказания медицинской помощи при проведении клинического исследования следует рассматривать с нескольких точек зрения:
А. Планируя клиническое исследование, как представители компании-спонсора, так и ответственные исследователи на каждой клинической базе должны определить, какая именно медицинская помощь и в каком объеме может понадобиться субъектам исследования в рамках данного клинического испытания (на основании детального анализа протокола) и заранее обеспечить возможность незамедлительного использования всех необходимых медицинских средств, если это понадобится;
Б. На этапе отбора исследовательских центров (то есть медицинских учреждений, где испытуемые будут проходить обследования и получать лечение в соответствии с протоколом исследования) сотрудники компании-спонсора или контрактной исследовательской организации должны удостовериться, что квалификация врача-исследователя и его помощников, врачей-соисследователей, соответствует задачам исследования. Другими словами, оценить, обладают ли члены исследовательского коллектива достаточными знаниями, опытом и навыками для того, чтобы выбирать пациентов или добровольцев для участия в исследовании, проводить необходимые лечебные и диагностические процедуры в соответствии с протоколом, а также своевременно диагностировать и лечить побочные явления, которые могут возникнуть в ходе исследования. Сюда же можно отнести и необходимость проверки наличия соответствующего технического оснащения в исследовательском центре (с другой стороны, ответственный исследователь и сам должен адекватно оценить силы и возможности своей команды, соглашаясь на то или иное исследование);
В. Еще во время подготовки клинического исследования и разработки информации для пациента и формы информированного согласия необходимо оценить, какую медицинскую помощь испытуемые могут получить в рамках этого испытания. Пациент может дать согласие на участие в клиническом исследовании ради получения более качественной и полной медицинской помощи, недоступной ему в рутинной клинической практике. Кроме дополнительной возможности получения лечения в случае тяжелого заболевания, это может быть, например, консультация ведущих специалистов, редкие и/или дорогостоящие методы обследования, подтверждение диагноза или выполнение анализов в лучших клиниках и лабораториях. В исследованиях, где не ожидается получение испытуемыми дополнительной пользы от лечения по сравнению с клинической практикой, это может быть возможность получить тщательное наблюдение за состоянием здоровья и уход, детальное обсуждение состояния испытуемого и выработку тактики лечения ведущими специалистами в данной клинической области, улучшение качества жизни. Когда пациент дал согласие на участие в клиническом испытании, ожидая получить более качественную и полную медицинскую помощь, исследователь должен сделать все возможное, чтобы оправдать его ожидания.
Мотивация испытуемых для участия в клиническом исследовании может быть основой для возникновения различных конфликтов, в том числе связанных с оказанием медицинской помощи. Согласно исследованию, проведенному путем опроса 1490 жителей различных европейских стран [3], желание получить более качественную медицинскую помощь было четвертым по распространенности мотивирующим фактором участия в исследованиях (48%), уступив место желанию помочь развитию медицинской науки (69%), дополнительному заработку (58%) и желанию помочь другим людям с подобной патологией (57%). Изданное в США подробное руководство для людей, рассматривающих возможность принять участие в клиническом исследовании [4, 5], не исключая положительных сторон для потенциальных испытуемых, достаточно жестко расставляет точки над «і»: «Часто лечение в качестве испытуемого клинического исследования ошибочно воспринимается так же, как получение медицинской помощи в качестве пациента. Реальность же заключается в том, что клинические исследования нацелены на получение ответов на научные вопросы, а не на лечение как таковое. Каждый желающий принять участие в клиническом исследовании должен понимать разницу между исследованием и обычным лечением». Таким образом, врач-исследователь при общении с испытуемым всегда должен помнить, о том, что у пациента или здорового добровольца существует своя точка зрения и свое восприятие происходящего.
При этом, однако, следует помнить, что в регионе развивающихся стран Центральной и Восточной Европы фактор возможности получения более качественной медицинской помощи может играть более важную роль, чем в США и Западной Европе, из-за меньшей доступности современных методов диагностики и лечения. То есть ожидания пациентов получить более качественную медицинскую помощь в рамках участия в клинических исследованиях нельзя недооценивать.
Как известно, основным документом, регламентирующим обязанности всех сторон-участниц клинического исследования, является руководство по надлежащей клинической практике (Good Clinical Practice — GCP) Международной конференции по гармонизации (International Conference on Harmonization — ICH) [6]. Вопросам оказания медицинской помощи испытуемым в этом документе посвящен только раздел 4.3, состоящий из четырех статей:
4.3.1. Квалифицированный врач (или стоматолог, в зависимости от ситуации), являющийся исследователем или соисследователем, должен нести ответственность за все решения по вопросам оказания медицинской помощи в рамках проводимого исследования.4.3.2. В течение и после окончания участия субъекта в исследовании исследователь/медицинское учреждение должны обеспечить оказание субъекту необходимой медицинской помощи в случае любых выявленных в ходе исследования негативных проявлений, включая клинически значимые отклонения лабораторных показателей. Исследователь/медицинское учреждение обязаны сообщить субъекту о необходимости лечения интеркуррентного(-ых) заболевания(-ий), выявленного(-ых) в ходе исследования.
4.3.3. Исследователю рекомендуется сообщить участковому врачу (лечащему врачу) об участии субъекта в испытании, если субъект наблюдается у такого врача и не возражает против уведомления последнего.
4.3.4. Хотя субъект испытания и не обязан сообщать о причинах, побудивших его досрочно прервать участие в испытании, исследователь должен попытаться выяснить эти причины, не нарушая при этом прав субъекта.
Рассмотрим эти положения более подробно.
ПУНКТ 4.3.1
Основой для этого положения является п. 15 Хельсинкской декларации («...Ответственность за испытуемого должна всегда лежать на специалисте с медицинской квалификацией и не может быть ответственностью самого испытуемого, несмотря на его согласие на участие в исследовании») [2]. Клиническое исследование может сопровождаться нестандартными (непредвиденными) ситуациями, что, безусловно, осложняет принятие правильных медицинcких решений, поэтому клиническими исследователями должны становиться врачи только с высокой профессиональной квалификацией. Средний и младший медицинский персонал ни при каких обстоятельствах не должен принимать медицински значимых решений при оказании медицинской помощи участникам клинического исследования.
Стоматолог в этом положении выделен отдельно, так как в некоторых странах профессия стоматолога является отдельной и не подразумевает обязательной квалификации «врач». В таких странах, по сути, эти специалисты делятся на стоматологов (dentist) и врачей-стоматологов. Однако стоматологи могут выступать в роли исследователей в определенных стоматологических клинических испытаниях. В Украине, России и других странах нашего региона все стоматологи имеют квалификацию врача и могут быть исследователями во всех профильных исследованиях .
Важно!
Несмотря на то что в данном пункте обозначены границы ответственности — «в рамках клинического исследования» — необходимо четко понимать, что они не ограничиваются только протоколом, а включают также все негативные проявления, возникшие у испытуемого во время участия в исследовании, а в некоторых случаях и после его окончания.
ПУНКТ 4.3.2
Обеспечение испытуемого необходимой медицинской помощью в случае развития у него любых побочных явлений, включая значимые изменения лабораторных и функциональных показателей, даже в тех случаях, когда такие изменения не сопровождаются клинической симптоматикой, является обязанностью врача-исследователя и медицинского учреждения, на базе которого проводится исследование. Если в ходе исследования у испытуемого будут выявлены сопутствующие заболевания, обязанность сообщить пациенту о таких заболеваниях и о необходимости их лечения (в тех случаях, когда лечение показано) возлагается на врача-исследователя и медицинское учреждение, на базе которого проводится исследование.
Важно!
Для каждого испытуемого «участие в исследовании» начинается с момента подписания им информированного согласия. Именно с этого момента на врача-исследователя возлагается ответственность за здоровье и благополучие пациента/здорового добровольца. Поэтому, даже если непредвиденное медицинское событие развилось до приема исследуемого препарата (то есть причинно-следственная связь исключена по определению), исследователь обязан предпринять необходимые меры для оказания испытуемому адекватной медицинской помощи. В случае, если активное участие исследователя невозможно (например больной госпитализирован в другом городе) или не необходимо (развившееся медицинское событие не входит в сферу компетенции исследователя), он должен поддерживать активный информационный обмен с лечащим врачом (при согласии больного или в случае, когда больной не давал отдельного согласия, но сам уже сообщил лечащему врачу об участии в исследовании)
ПУНКТ 4.3.3
В случае, когда испытуемый постоянно наблюдается у одного и того же врача (например, участкового терапевта по месту жительства, семейного врача, участкового онколога в связи с основным заболеванием и т.д.), рекомендуется сообщать ему об участии в клиническом исследовании (с указанием группы исследуемого препарата и основного показания к назначению). Во-первых, именно такой специалист может первым отметить изменения в состоянии здоровья больного (как в лучшую, так и в худшую стороны), а значит, и обратить на них внимание врача-исследователя или самого больного. А, во-вторых, в таком случае будет возможно согласовать тактику ведения больного, включая назначение или не-назначение определенных препаратов сопутствующей терапии, не-повторение одних и тех же диагностических процедур (например ежегодного рентгена легких/флюорографии, регулярной коагулограммы и т.д.) и др. Однако необходимо помнить о праве пациента/здорового добровольца на сохранение в тайне его личной информации. Поэтому обсуждать с лечащим врачом участие больного в исследовании следует только после его согласия на такое обсуждение.
ПУНКТ 4.3.4
В том случае, если испытуемый досрочно прерывает участие в клиническом исследовании, исследователю настоятельно рекомендуется выяснить причины такого решения. Несмотря на то что испытуемый не обязан предоставлять обоснование такого решения (как правило, это прописано и в информации для участника клинического исследования), исследователь должен приложить все усилия, чтобы получить такую информацию. Дело в том, что кроме личных причин (переезд в другой город, непонимание со стороны родственников, недоверие своему врачу, увиденная «обличительная» передача по телевидению и др.), отказаться от дальнейшего участия в исследовании пациента могут побудить неблагоприятные медицинские проявления, которые сам больной может и не связывать с приемом исследуемого препарата (например депрессия или повышенная утомляемость) или по каким-то причинам стесняться говорить о них (к примеру геморрой, снижение потенции, выделения с неприятным запахом и др.). Чрезвычайно важно получать информацию о таких побочных явлениях, чтобы последующий анализ данных, а следовательно, и вывод об эффективности и/или безопасности препарата был максимально точен.
Вопрос-ответ
1. Какие требования ICH GCP, связанные с оказанием медицинской помощи испытуемым, обязана выполнить компания-спонсор на стадии подготовки исследования?
Согласно требованиям ICH GCP (п. 5.4.1) еще на стадии подготовки к исследованию спонсор обязан привлечь квалифицированных специалистов — врачей и фармакологов, для того чтобы обеспечить научное обоснование исследования, соответствие его программы поставленным медицинским задачам и безопасное проведение исследования.
2. Какие элементы протокола исследования регламентируют оказание оптимальной медицинской помощи испытуемым?
Уже на этапе разработки протокола исследования в него должен быть заложен перечень обследований, которые необходимо будет выполнить испытуемому перед началом исследования и в ходе его проведения. Он должен включать обследования, необходимые как для оценки эффективности исследуемого препарата, так и для своевременного выявления его неблагоприятного воздействия на организм испытуемого (тем самым повышая безопасность исследования). Перечень необходимо составить таким образом, чтобы обследование было максимально эффективным и стандартизованным, и в то же время минимально неприятным и травматичным для испытуемого. Кроме того, необходимо составить программу наблюдения за испытуемым, включая регулярные медицинские осмотры врачом-исследователем и лабораторный и инструментальный мониторинг состояния испытуемого.
3. Как влияет знание протокола исследования врачами-исследователями на качество оказания медицинской помощи испытуемым? Обязан ли ответственный исследователь и соисследователи детально ознакомиться со всеми положениями протокола исследования?
Ст. 4.1.2 и 4.2.4 ICH GCP требуют от ответственного исследователя и других членов исследовательского коллектива хорошего знания протокола и других предоставленных спонсором материалов исследования. Это означает, что спонсор обязан не просто предоставить членам исследовательского коллектива протокол и другие документы исследования, но и провести для них соответствующее обучение до начала исследования.
Как правило, основное обучение материалам предстоящего исследования проводится в рамках стартового совещания исследователей (которое еще называют встречей исследователей). Совершается большая ошибка, если ответственные исследователи по каким-либо причинам отказываются от посещения таких совещаний и делегируют эту обязанность кому-либо из соисследователей. Это приводит к тому, что к моменту начала исследования ответственные исследователи не имеют четкого представления о его основных документах и процедурах, а, следовательно, качество проведения клинического испытания на клинической базе под руководством такого исследователя оказывается существенно ниже, чем в центре исследователей, прошедших обучение в рамках стартового совещания. Недостаточная вовлеченность ответственного исследователя, наиболее квалифицированного специалиста в исследовательской команде, и/или недостаточное знание им основных документов исследования, в том числе содержащих подробную информацию об исследуемом препарате, может привести к тому, что исследователь не сможет вовремя выявить развитие побочного явления и оказать необходимую помощь пациенту. Несвоевременная и неправильная регистрация развивающихся у испытуемых побочных явлений, кроме повышения риска для испытуемых, ведет к неправильному предоставлению спонсору исследования, а через него и регуляторным органам, данных по безопасности исследуемого препарата.
4. Какие требования предъявляет руководство ICH GCP к квалификации врача-исследователя?
П. 4.1.1 ICH GCP содержит прямое требование привлекать к участию в испытаниях квалифицированных врачей-исследователей. Квалификация врача-исследователя должна быть подтверждена соответствующим медицинским образованием, специальной дополнительной подготовкой (специализацией) и опытом работы. Следует ли считать, что только профессор или научный сотрудник с ученой степенью отвечает таким требованиям? Конечно нет. Квалифицированный и опытный врач или заведующий отделением, который прошел необходимую специализацию и имеет большой и постоянно растущий опыт работы с целевой категорией пациентов, нередко является лучшим из возможных исследователей, так как, постоянно работая с пациентами в медицинском учреждении, именно он имеет возможность уделять испытуемым необходимое время.
5. Всегда ли сотрудник с научной степенью является лучшим из возможных исследователей?
Проведение клинического испытания, конечно, относится к научно-исследовательской сфере медицинской деятельности, но не следует путать непосредственные функции врача-исследователя с обработкой данных исследования и их научным анализом. От врача-исследователя требуется, прежде всего, каждодневное и квалифицированное обследование и лечение пациентов в соответствии с протоколом исследования. Чрезвычайно важно, чтобы исследователь и члены исследовательской команды были доступны пациенту в любое время суток, для того чтобы оказать медицинскую помощь в случае необходимости. На это, в числе прочего, указывают п. 4.2.2 и 4.2.3 действующей редакции ICH GCP. Таким образом, практикующие врачи, наряду с научными сотрудниками и преподавателями медицинских вузов, должны шире привлекаться к проведению клинических исследований. Это же относится и к медицинским учреждениям. Многопрофильные больницы, равно как и специализированные клиники (муниципальные и частные), могут эффективно использоваться в качестве клинических баз для проведения клинических исследований (см. ICH GCP, глава «Терминология», п. 1.59).
6. В каких дополнительных документах спонсор и исследователь/медицинское учреждение оговаривают вопросы оказания медицинской помощи испытуемым?
Важный аспект проведения клинического исследования — заключение соглашения/договора между спонсором и исследователем и/или медицинским учреждением. Заключение соглашения/договора является требованием ICH GCP (п. 5.1.4 и 5.6.3) и должно производиться до начала исследования. В таком соглашении медицинские учреждения, в частности, могут оговорить возможность компенсации со стороны спонсора дополнительных расходов, которые могут возникнуть у учреждения в случае необходимости оказания испытуемым дополнительной медицинской помощи.
7. Откуда исследователи и регуляторные органы получают информацию о безопасности исследуемого препарата?
Перед началом исследования спонсор обязан предоставить исследователям полную информацию о безопасности и эффективности исследуемого препарата. В дальнейшем, в ходе исследования, вся вновь поступающая информация о безопасности исследуемого препарата должна немедленно передаваться исследователям (руководство ICH GCP, п. 5.12.1 и 5.12.2). При этом, если в ходе исследования спонсору становится известно о фактах, которые могут неблагоприятно повлиять на безопасность испытуемых или их согласие на продолжение участия в исследовании (например развитие выраженных негативных проявлений у других испытуемых в данном проекте или в иных исследованиях с использованием данного препарата), спонсор обязан незамедлительно предоставить эту информацию не только исследователям, но также и в регуляторные органы и комитеты по вопросам этики всех стран, в которых проходят клинические испытания данного исследуемого препарата (ICH GCP, п. 5.16.2). Обязательные ежегодные отчеты о безопасности при продолжительных исследованиях, которые должен предоставлять спонсор регуляторным органам и этическим комитетам, очень важны, поскольку своевременный анализ информации обо всех побочных явлениях, частоте их возникновения и возможности взаимосвязи с применением исследуемого лекарственного средства позволит при необходимости своевременно прекратить/приостановить клиническое исследование и, соответственно, предотвратить развитие схожих побочных реакций у других испытуемых.
8. Как определить лечебную тактику при развитии угрожающих жизни серьезных побочных явлениях в случае двойного слепого дизайна клинического исследования?
Для того чтобы грамотно разобраться в подобной ситуации, исследователь еще до включения в клиническое исследование испытуемых должен тщательно изучить предложенные им документы (включая протокол исследования и брошюру исследователя) и найти в них ответы на следующие вопросы:
1. Каков предполагаемый или известный механизм действия исследуемого препарата?
2. Какие серьезные побочные реакции уже отмечали в ходе клинической разработки исследуемого лекарственного средства и какое лечение применяли?
3. Выявляли ли побочные реакции, которые при усугублении могли бы стать жизнеугрожающими (например кратковременный приступ стенокардии не является серьезным побочным явлением, но инфаркт миокарда, осложненный кардиогенным шоком, уже является таковым), с какой частотой?
4. Существует ли специфический антидот к исследуемому препарату (или специфическое лечебное вмешательство), доступно ли оно исследователю, какова процедура его введения (или реализации вмешательства)?
5. Какую процедуру раскрытия рандомизационного кода предусматривает протокол клинического исследования и что конкретно сказано по поводу действий исследователя в неотложных ситуациях?
6. Есть ли существенные отличия в механизме действия (и соответственно предполагаемых побочных реакциях) между исследуемым препаратом и препаратом сравнения?
Возможно, документы исследования содержат достаточное количество информации, которое позволит исследователю правильно поступить в любой сложной ситуации, всего лишь следуя разработанным для него инструкциям. Несогласие с предложенными инструкциями целесообразно высказывать до начала клинического исследования, а не во время развития неотложной ситуации, но в конечном итоге ответственность за здоровье испытуемого всегда лежит на исследователе, следовательно, и окончательные клинические решения принимать должен исследователь и это решение всегда должно быть принято в интересах пациента. Однако в некоторых случаях имеющейся в документах исследования информации может быть недостаточно (например в связи с ранней фазой клинической разработки и малым количеством накопленных данных, с упущениями при написании таких документов и др.). В таких случаях исследователю рекомендуется действовать с учетом алгоритма (рисунок).
|
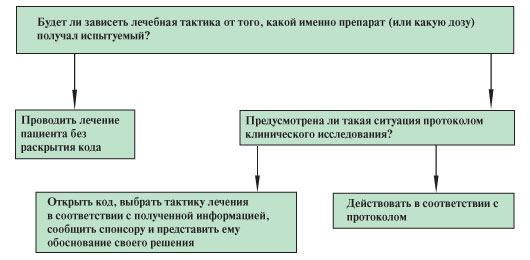
Таким образом, ставя интересы испытуемого, заботу о его здоровье и благополучии на первое место, врач должен помнить о своих обязанностях исследователя и стремиться максимально придерживаться протокола (в частности, не раскрывать рандомизационный код без достаточных на то оснований). Важность сохранности ослепления кодов лечения для исследования в целом обусловлена требованиями к статистической значимости и борьбе с предвзятостью на всех этапах, что было более подробно описано в статье, посвященной рандомизации в клинических исследованиях. n
СПИСОК ЛИТЕРАТУРЫ | |
|
Максим Белоцерковский, Ольга Голубева, Ирина Тесленко, Ольга Миронова, Елена Руднева