.png)
Доказательная медицина: новое в поиске доказательств
- Публикации /
-
3287
АННОТАЦИЯ
Обзор посвящён обсуждению новых направлений в доказательной медицине, переосмыслению доказательной базы в медицине с упором на демонстрацию эффективности и безопасности новых терапевтических средств и медицинских технологий, от редактирования генов до алгоритмов искусственного интеллекта. В то же время клинические исследования лекарственных средств (ЛС) тоже не стоят на месте, происходит активное развитие, появляются новые подходы, методы и дизайны. Процесс разработки, регистрации и выхода на рынок любого ЛС занимает значительный период времени, требует высоких финансовых затрат и человеческих ресурсов. Золотым стандартом доказательной медицины исходно считали рандомизированные контролируемые исследования, в то же время для них присущи объективные недостатки: ограниченность выборки, неспособность контролировать все факторы; недостаточное время наблюдения, негативное воздействие на участников, невозможность определить причинно-следственные связи; ограниченность методов измерения, ограниченный бюджет и др. В данном обзоре рассматриваются направления развития методологии клинических исследований. Активное внедрение новых дизайнов, таких как Адаптивные клинические испытания, «Мастер» протоколы, включающие Зонтичное исследование, Корзинное исследование, Исследования платформы, Мастер-обсервационные испытания и расширение использования Испытаний на одном пациенте (N-of-1 trial), позволяет сделать исследования более эффективными, релевантными и применимыми к реальным условиям практики здравоохранения и пациентоориентированными. В обзоре мы также рассматриваем как положительные, так и отрицательные стороны расширенного применения биомаркеров и Цифровых конечных точек в клинических исследованиях и виртуальных клинических испытаний, в которых используют математические модели для изучения гетерогенности пациентов и её влияния на решение различных терапевтических вопросов. Процесс экспертизы и регуляторного одобрения по-прежнему замедляет выход ЛС на фармацевтический рынок. В обзоре рассматриваются изменения в подходах регуляторов (FDA, EMEA и Минздрав России) к процедуре регистрации ЛС. Пандемия COVID-19 и санкции привели к необходимости расширения списка ЛС с разрешённой ускоренной регистрацией. Однако ускоренная регистрация вызывает много вопросов, касающихся достаточности уровня доказательности и безопасности. Как мы видим, необходим глубокий синтез и объединение всех имеющихся данных для достижения доказательной медицины следующего поколения. Основная задача в ближайшие два десятилетия будет заключаться в том, чтобы использовать потенциал многомерной генерации доказательств путём извлечения, сопоставления и получения больших массивов данных естественного течения заболеваний, геномики и всех других анализов омики, всех опубликованных клинических исследований, RWD для предоставления доказательств следующего поколения.
ВВЕДЕНИЕ / INTRODUCTION
Доказательная медицина (англ. Evidence-based medicine; ДМ) — это научный подход к медицинской практике, основанный на анализе надёжности и достоверности клинических исследований [1]. Современная медицина опирается на фактические данные, но это относительно недавнее явление. Первый мета-анализ был проведён в 1904 г. для оценки эффективности прививок против брюшного тифа, а первые рандомизированные контролируемые исследования (РКИ) были опубликованы в 1940-х годах [2]. Устоявшийся термин «доказательная медицина» появился в 1992 году, мета-анализы и систематические обзоры составили основу этой области и сформировали основу для создания клинических рекомендаций [3].
Актуальность применения ДМ в настоящее время объясняется несколькими факторами. Во-первых, быстрый темп развития науки и технологий: появляются новые диагностические и лечебные методы, ДМ может гарантировать, что они действительно работают и не наносят вреда пациентам. Развитие технологий в генном, иммунологическом, протеомном, метаболическом, микробном и вирусном профилях стали двигателями этих достижений. «Массовая демонстрация» машинного обучения — использование алгоритмов для просеивания больших молекулярных, биохимических и геномных данных. Помимо этого, в клинической практике стали применять такие направления, как:
- оценка технологий здравоохранения (англ. Health Technology Assessment; ОТЗ): эта область включает в себя анализ не только ЛС, но и технологий, таких как телемедицина, медицинские приборы, методы хирургического вмешательства и другие;
- искусственный интеллект (англ. Аrtificial intelligence; ИИ): использование машинного обучения и алгоритмов в области медицинских исследований позволяет эффективно обработать большие объёмы данных и получить новые знания о различных заболеваниях;
- данные реальной клинической практики (англ. real-world data; RWD) и доказательства, полученные на основе анализа данных реальной клинической практики (англ. real-world evidence; RWE): широкое применение как при доклиническом и клиническом этапе изучения ЛС, так и при регистрации.
Во-вторых, увеличение числа пациентов с орфанными заболеваниями: необходима строгая система оценки результатов лечения таких пациентов. В-третьих, увеличение стоимости здравоохранения: ДМ помогает выбрать наиболее экономически целесообразные методы и лекарственные средства (ЛС).
В январе 2023 года журнал «Nature Medicine» запустил новую тематическую серию статей «Rethinking evidence in medicine» («Переосмысление доказательств в медицине») [4]. Раздел посвящён обсуждению новых направлений в ДМ, а в редакционной статье авторы обосновывают необходимость выпуска серии: «Технологический прогресс требует новых подходов, поэтому сегодня мы начинаем новую серию статей о переосмыслении доказательной базы в медицине с упором на демонстрацию эффективности и безопасности новых терапевтических средств и медицинских технологий, от редактирования генов до алгоритмов ИИ» [5]. В то же время клинические исследования ЛС тоже не стоят на месте, происходит активное развитие, появляются новые подходы, методы и дизайны.
ПРОБЛЕМЫ КЛАССИЧЕСКИХ ПРЕДРЕГИСТРАЦИОННЫХ ИНТЕРВЕНЦИОННЫХ КЛИНИЧЕСКИХ ИСПЫТАНИЙ / PROBLEMS OF CLASSICAL PRE-REGISTRATION INTERVENTIONAL CLINICAL TRIALS
Процесс разработки, регистрации и выхода на рынок любого ЛС занимает значительный период времени, требует высоких финансовых затрат и человеческих ресурсов. На рис. 1 представлена классическая временная шкала от разработки до первых клинических испытаний на людях и в конечном счёте одобрения регулятором [3][6][7]. Золотым стандартом доказательной медицины исходно считали РКИ, в то же время для них присущи объективные недостатки:
- ограниченность выборки: стандартные дизайны часто используют выборки, которые не являются представительными для целевой популяции. Это может привести к ошибочным выводам и недостаточной обобщаемости результатов. Недостаточное количество пациентов в исследовании, что может снизить мощность статистических тестов и повысить вероятность ложноотрицательных результатов;
- неспособность контролировать все факторы: стандартные дизайны не всегда позволяют надёжно контролировать все потенциальные факторы, которые могут влиять на результаты. Это может привести к искажениям результатов и искажению выводов;
- недостаточное время наблюдения: многие стандартные дизайны имеют ограниченное время наблюдения, что может повлиять на способность исследования отслеживать долгосрочные результаты. Короткий период исследования может быть недостаточным и не позволит оценить полный спектр эффектов нового препарата или терапии, особенно если речь идёт о медленно прогрессирующих заболеваниях;
- негативное воздействие на участников: некоторые стандартные дизайны могут негативно влиять на участников исследования, например, заставляя их получать ненужное лечение или подвергая их другим рискам;
- невозможность определить причинно-следственные связи: многие стандартные дизайны не позволяют надёжно определить причинно-следственные связи между переменными, что может затруднить интерпретацию результатов;
- ограниченность методов измерения: стандартные дизайны могут ограничивать способы измерения переменных, что может снижать точность и достоверность результатов;
- ограниченный бюджет: отведённый бюджет может также ограничить возможности проведения более масштабных исследований, которые могут быть более репрезентативными и показательными.
Как видно из данных, представленных на рис. 1, можно в текущем подходе изучения ЛС визуализировать временную шкалу от разработки ЛС до первых испытаний на людях и в конечном счёте одобрения регулятором [3][6]. Будущий подход подразумевает широкое применение ИИ, данных электронных медицинских карт, интернет-медицинских вещей, глубокой нейронной сети и машинного обучения.

Рис. 1. Хронология изучения лекарственного препарата: от настоящего к будущему
Fig. 1. Chronology of the drug study: from present to future
Примечания: Адаптировано по Subbiah V (2023) [6]: AI — искусственный интеллект; DNN — глубокая нейронная сеть; HER — электронные медицинские карты; IoMT — интернет медицинских вещей; ML —машинное обучение.
Notes: Adapted by Subbiah V (2023) [6]: AI — Artificial intelligence; DNN — Deep Neural Network; HER — Electronic Health Record; IoMT — Internet of Medical Things; ML — Machine Learning.
РАЗВИТИЕ МЕТОДОЛОГИИ КЛИНИЧЕСКИХ ИССЛЕДОВАНИЙ / DEVELOPMENT OF CLINICAL RESEARCH METHODOLOGY
В последние годы в парадигме клинических исследований наблюдают активное внедрение новых дизайнов.
АДАПТИВНЫЕ КЛИНИЧЕСКИЕ ИСПЫТАНИЯ / ADAPTIVE DESIGN CLINICAL TRIALS
Для преодоления существенных недостатков классических дизайнов в начале 2000-х годов были предложены и проведены первые исследования с «адаптивным дизайном». В 2010 г. были выпущены рекомендации Управления по санитарному надзору за качеством продуктов и медикаментов США (англ. Food and Drug Administration; FDA) с правилами проведения таких исследований [8]. Адаптивный дизайн определяется как дизайн клинического исследования, который позволяет адаптировать, модифицировать необходимые аспекты дизайна исследования после его инициации и запуска без потери значимости, целостности, адекватности и обоснованности исследования. Адаптивный дизайн включает проспективно запланированную возможность модификации одного или более элементов дизайна и гипотез на основе анализа полученных данных (обычно данных промежуточного анализа). Это означает, что все изменения планируются заранее, детали определяются как можно раньше и до того, как разослеплённые данные могут стать доступны специалистам, участвующим в планировании адаптаций [8]. Эти модификации могут ускорить процесс разработки ЛС или использоваться для более эффективного распределения ресурсов без снижения научных и регуляторных стандартов. Определение адаптивного дизайна накладывает существенные ограничения, и в то же время оно позволяет проводить достаточно широкий спектр адаптаций: пересматривать критерии отбора (выбор популяции, для которой терапия имеет наибольшее преимущество), размер выборки и/или длительность исследования, параметры рандомизационной процедуры, режимы дозирования (дозовые уровни, схема, длительность и т. п.), число терапевтических групп, сопутствующую терапию, схему оценки состояния пациента, первичную конечную точку (например, единичная (unitary) против композитной (composite), компоненты композитной, временные моменты оценки), вторичные конечные точки (выбор, порядок тестирования), методы анализа данных (ковариаты для финального анализа, статистическая методология, контроль ошибки первого рода) [9].
Одним из основных рисков исследований с адаптивным дизайном является «операционное смещение» (англ. operational bias), возникающее при раскрытии кода рандомизации во время промежуточного анализа, т. е. промежуточный результат может стать известен исследователям и, как следствие, повлиять на поведение исследователей и вызвать смещение. Важно обеспечить сохранение ослепления команды, в связи с этим в целях получения не ангажированного мнения решение часто принимается после рассмотрения результатов Независимым Комитетом по Безопасности Данных (англ. Independent Data/Safety Monitoring Boards) [6].
К типичным проблемам адаптивного дизайна относят то, что некорректные результаты могут быть обнаружены во время промежуточного анализа данных, проведённого в неправильно выбранный момент времени. Как следствие, есть риск «закрытия» некоторых якобы неэффективных терапевтических групп. Это может также привести к неадекватному выявлению проблем с безопасностью, что является одной из основных целей исследования. Адаптивный дизайн может также несколько «сдвинуть» целевую популяцию пациентов, что приведёт к ограниченной генерализации полученных результатов. Потенциальным недостатком адаптивного дизайна является возможность переоценивать эффект терапии. Например, если адаптивный дизайн выбирает наилучший из наблюдаемых на промежуточном анализе эффектов терапии, который может быть проявлением случайности, то такая оценка эффекта будет превышать его реальный размер [6].
«МАСТЕР» ПРОТОКОЛЫ / MASTER PROTOCOL
За последнее десятилетие достигнут существенный прогресс в разработке, проведении и внедрении «мастер» протоколов — всеобъемлющих протоколов, которые применяются к нескольким подисследованиям [10]. Под мастер-протоколами в клинических испытаниях понимается тип дизайна исследования, позволяющий изучать несколько исследовательских вопросов или вмешательств в рамках одного общего протокола. В отличие от традиционных клинических испытаний, в которых основное внимание уделяется одному вмешательству или исследовательскому вопросу, мастер-протоколы позволяют оценить несколько методов лечения или гипотез одновременно. Мастер-протоколы могут включать параллельные интервенционные исследования при одном заболевании или нескольких заболеваниях, определяемых биомаркером или нозологией заболевания [11]. Существует несколько типов таких протоколов: зонтичное исследование, корзинное исследование, исследования платформы и мастер-обсервационные испытания (рис. 2). Каждый из них представляет собой уникальную конструкцию испытания, которая может включать в себя независимый вид вмешательства с контрольными видами вмешательства и может анализироваться индивидуально и/или коллективно, с дополнительной гибкостью [10][12]. Наибольшее применение такие дизайны нашли в онкологии, благодаря достижениям в геномике (выявление молекулярных изменений), разработке ЛС и быстрой клинической трансляции, что открывает эру прецизионной онкологии.
Мастер-протоколы обладают рядом преимуществ по сравнению с традиционными схемами клинических испытаний. Они позволяют более эффективно использовать ресурсы и сокращать сроки, поскольку одновременно можно проводить несколько видов лечения или решать несколько исследовательских вопросов. Кроме того, мастер-протоколы облегчают проведение адаптивных испытаний, позволяя исследователям вносить изменения в протокол исследования на основе накопленных данных. Однако у мастер-протоколов есть целый ряд проблем — координация между различными лечебными группами или подгруппами может быть сложной и требует тщательного статистического планирования для учёта возможных погрешностей или сбивающих факторов. В целом мастер-протоколы способны ускорить разработку новых методов лечения и улучшить результаты лечения пациентов за счёт эффективной оценки нескольких вмешательств или исследовательских вопросов в рамках одной схемы исследования.

Рис. 2. Классы мастер-протоколов
Fig. 2. Master protocol classes
1. ЗОНТИЧНОЕ ИССЛЕДОВАНИЕ / UMBRELLA TRIAL
Зонтичные испытания предполагают оценку нескольких различных методов терапии или подходов к лечению в рамках одного заболевания или состояния. Участники делятся на различные подгруппы по определённым характеристикам или биомаркерам, и каждая подгруппа получает различное лечение. Примерами таких исследований могут быть исследования рака молочной железы I-SPY (Investigation of Serial Studies to Predict Your Therapeutic Response With Imaging And Molecular Analysis) [13] и рака лёгкого Lung-MAP (Biomarker-driven therapies for previously treated squamous non-small-cell lung cancer) [14].
2. КОРЗИННОЕ ИССЛЕДОВАНИЕ / BASKET TRIAL
Эти испытания часто применяют в онкологии, предполагают тестирование конкретной терапии на нескольких различных типах опухолей, как правило, имеющих общие молекулярные изменения или пути развития. Цель — выявить потенциальные специфические генетические изменения или мишени, которые могут ответить на конкретную терапию. Например, исследование VE-Basket (VE, вемурафениб) [15], исследование ROAR (Rare Oncology Agnostic Research, агностическое исследование редких онкологических заболеваний) [16] и исследование ARROW [17].
3. ИССЛЕДОВАНИЯ ПЛАТФОРМЫ / PLATFORM TRIAL
Это многогрупповые, многоэтапные дизайны исследований, по которым сравнивают несколько лечебных групп с общей контрольной группой в контексте одного и того же мастер-протокола. Эти испытания предназначены для непрерывной оценки нескольких исследуемых вмешательств или схем лечения конкретного заболевания. В ходе испытания могут быть добавлены или отменены новые методы лечения, что позволяет вносить адаптивные изменения на основе текущего анализа данных. Кроме того, они могут быть вечными / бессмертными (без определённой даты окончания) и быть более эффективными по сравнению с традиционными исследованиями благодаря наличию общей контрольной группы, что обеспечивает включение большей доли пациентов в интервенционные / экспериментальные группы, чем в контрольную группу. В качестве примера можно привести платформенное исследование RECOVERY (Randomised Evaluation of COVID-19 Therapy) [18]. Исследования платформы являются гибкими по своей конструкции и не обязательно должны иметь общую контрольную группу. Основная идея заключается в том, что интервенционные группы могут быть добавлены к текущему испытанию, например, как в исследовании платформы plasmaMATCH (The UK Plasma Based Molecular Profiling of Advanced Breast Cancer to Inform Therapeutic Choices, Молекулярное профилирование распространённого рака молочной железы на основании плазмы для информирования о выборе видов терапии в Великобритании) [19]. Хотя вышеупомянутые испытания были разработаны в контексте разработки ЛС в области онкологии и инфекционных заболеваний, возможности платформенных испытаний могут быть использованы и в других различных областях, таких как клиническая психология и неврология. Такие испытания могут также использоваться для цифровых вмешательств в области психического здоровья и могут быть легко реализованы в условиях ограниченных ресурсов [20].
МАСТЕР-ОБСЕРВАЦИОННОЕ ИСПЫТАНИЕ / (MASTER OBSERVATIONAL TRIAL; МОТ)
MOT представляет собой перспективный дизайн наблюдательного исследования, при котором набор пациентов проводится широко, независимо от сигнатуры биомаркеров, и собирают исчерпывающие данные о каждом участнике [12][21]. MOT представляет собой сочетание мастер-интервенционного исследования, и проспективных наблюдательных испытаний, и попыток гибридизации мощности мастер-интервенционных протоколов на основе биомаркеров с широтой данных реальной клинической практики (RWD). Этот подход может быть хорошо приспособлен для сбора потенциальных RWD по многим специальностям. Одним из примеров мастер-обсервационного испытания является реестр исходов при онкологических заболеваниях ROOT (англ. Master Registry of Oncology Outcomes Associated with Testing and Treatment) [12].
ИСПЫТАНИЯ НА ОДНОМ ПАЦИЕНТЕ / N-OF-1 TRIAL
При определении оптимального лечения у пациента традиционные испытания подвержены предвзятости. Крупномасштабные РКИ могут служить лишь частичным ориентиром, а для большинства клинических заболеваний они не проводились или не могут быть проведены. Однако РКИ на отдельных пациентах (N-of-1 trial) в некоторых случаях могут стать решением этой дилеммы. В N-of-1 trial пациент проходит пары периодов лечения (один период в каждой паре с активным препаратом и один с подобранным плацебо, назначаемым случайным образом); пациент и врач слепы к распределению, а цели лечения контролируются. N-of-1 trial целесообразно проводить при хронических стабильных заболеваниях, для лечения которых предлагаемый препарат, обладающий быстрым началом действия и прекращающий его вскоре после завершения, показал свою эффективность в открытом испытании терапии. Мониторинг целей лечения обычно включает количественное измерение симптомов пациента с помощью простых дневников или опросников. Пары периодов лечения продолжаются до тех пор, пока эффективность не будет доказана или опровергнута [6]. Ещё на заре доказательной медицины Guyatt G и Sackett D (1988) выпустили рекомендации для врачей по проведению таких исследований [22]. В эпоху индивидуализированной геномной медицины и развития испытания на одном пациенте становятся инструментом для изучения потенциально смертельных редких заболеваний. В качестве примера можно привести терапию антисмысловым олигонуклеотидом. Она была разработана и оценена у одного пациента, у которого было смертельное генетическое нейродегенеративное расстройство, известное как нейрональный цероидный липофусциноз CLN7 (форма болезни Баттена) [23].
N-of-1 trial имеют существенные риски:
- ограниченная репрезентативность: испытание может не дать полной картины о реакции других пациентов на новое ЛС или метод лечения. Результаты могут быть непредсказуемыми и не отражающими реальной практики использования в клинической практике;
- малая выборка: исследования имеют очень ограниченную выборку, что может ограничить статистическую значимость и репрезентативность полученных результатов. Их интерпретация может быть сложной из-за малого размера выборки;
- риск нежелательных эффектов: испытания могут быть более рискованными, так как нет возможности исследовать и предвидеть нежелательные эффекты наложения разных ЛС или методов лечения. Это может привести к потенциально опасным последствиям для пациента;
- непредсказуемость долгосрочных результатов: испытания могут быть недостаточными для оценки долгосрочной эффективности и безопасности нового ЛС или метода лечения. Необходимы исследования на большей выборке для более точной оценки долгосрочных результатов.
РАЗРАБОТКА БИОМАРКЕРОВ / BIOMARKER DEVELOPMENT
Исследования, проводимые по мастер-протоколам, требуют выявления биомаркеров с доказанной связью с исходами (клинические, патологические или физиологические), и контекст их использования для каждого процесса заболевания, и очерчивание чётких конечных точек для исследований. Разработка биомаркеров способствовала прогрессу в дизайне клинических испытаний, а беспрецедентные достижения в области геномики и иммунологии привели к тому, что в последнее десятилетие было одобрено несколько таргетных препаратов и иммунотерапий на основе биомаркеров [24][25]. Биомаркеры могут быть диагностическими, прогностическими, могут служить основой для ранней разработки ЛС, выбора дозы и дизайна испытаний. Однако уровень доказательности того или иного биомаркера во многом зависит от контекста его использования.
ЦИФРОВЫЕ КОНЕЧНЫЕ ТОЧКИ / DIGITAL ENDPOINTS
Это устройства или программные приложения, которые используются для сбора, мониторинга, анализа и обработки медицинских данных, такие как использование микрофонов смартфонов для мониторинга ухудшения когнитивной функции у людей с болезнью Альцгеймера или мониторов умных часов для оценки эффекта ЛС у людей с серповидноклеточной анемией [6][26]. C увеличением децентрализованного проведения испытаний по многим специальностям дистанционный мониторинг будет расширяться. Например, в недавнем исследовании была разработана модель ИИ для обнаружения и отслеживания прогрессирования болезни Паркинсона (для которой нет биомаркеров) на основе ночных дыхательных сигналов с использованием неинвазивной оценки на дому, предоставляя доказательства того, что ИИ может быть полезен в оценке риска доклинической диагностики состояния [26]. Цифровые конечные точки позволяют более достоверно оценить опыт пациента, раскрывают ранее невыразимые реалии бремени болезни и позволяют вдвое сократить затраты на разработку ЛС. Однако, прежде чем эти преимущества будут реализованы, необходимо приложить усилия не только к техническому созданию цифровых конечных точек, но и к созданию среды для их разработки и применения. Будущее цифровых конечных точек зависит от значимого междисциплинарного сотрудничества, достаточного количества доказательств того, что цифровые конечные точки могут реализовать свои перспективы, а также от создания экосистемы, в которой можно будет анализировать огромные объёмы данных, генерируемых цифровыми конечными точками [26]. Цифровая характеристика и оценка клинического статуса должны быть стандартизированы и гармонизированы с междисциплинарным сотрудничеством и вкладом. Консенсус также необходим для выявления и характеристики промежуточных и суррогатных конечных точек для основных хронических заболеваний. Это требует специализированного включения нескольких уровней данных, таких как геномные, протеомные и основанные на генотипе клинические данные и измерения по конкретным заболеваниям, в дополнение к слою функциональных данных. Национальный институт здравоохранения США (англ. The National Institutes of Health; NIH) и FDA разработали ресурс BEST (Biomarkers, EndpointS and other Tools; биомаркеры, конечные точки и другие инструменты) для прояснения неопределённости в отношении биомаркеров и конечных точек. Это «живой документ», который постоянно обновляется по мере изменения стандартов и доказательств, и разъясняет важные определения, и описывает некоторые иерархические отношения, связи и зависимости между терминами. На данный момент в России аналогичных общедоступных ресурсов не разработано. Однако использование цифровых конечных точек в медицине также сопровождается рисками и проблемами, такими как защита данных пациентов, конфиденциальность и этические вопросы.
ИЗМЕНЕНИЯ В ПРОЦЕССЕ ПРОВЕДЕНИЯ КЛИНИЧЕСКИХ ИССЛЕДОВАНИЙ / CHANGES IN THE PROCESS OF CONDUCTING CLINICAL TRIALS
Компонентами проведения клинических исследований являются реализация протокола; подбор, набор, мониторинг пациентов; обеспечение соблюдения требований по отчётности по безопасности, мониторинг; сбор и анализ данных. На фоне пандемии коронавирусной инфекции клинические испытания начали постепенно трансформироваться (см. рис. 1) [27]. В частности, это привело к децентрализованным испытаниям, цифровым, удаленным и «виртуальным» испытаниям (которые позволяют пациентам получать доступ к испытаниям независимо от их географического местоположения), а также к формированию концепции «больницы на дому» [28].
ВИРТУАЛЬНЫЕ КЛИНИЧЕСКИЕ ИСПЫТАНИЯ / VIRTUAL TRIALS
В последнее время всё большую популярность приобретают виртуальные клинические испытания, в которых используют математические модели для изучения гетерогенности пациентов и её влияния на решение различных терапевтических вопросов [6]. Виртуальные клинические испытания осуществляются без физического посещения медицинской организации. Они могут включать удалённый сбор данных, консультации через видеосвязь и использование мобильных устройств для самооценки состояния пациентами. Это может быть особенно полезно в ситуациях, когда физическое присутствие пациентов ограничено или неудобно. В течение последних десятилетий математическое моделирование играло ключевую роль в процессе разработки и анализа использования ЛС. В настоящее время модерирование способно оказать дальнейшую поддержку этому процессу с помощью новых методов клинических испытаний in silico (эксперимент, проведённый на компьютере или с помощью компьютерного моделирования) и «цифровых двойников». Такие клинические испытания могут позволить сделать процесс разработки лекарств более финансово эффективным, безопасным и эффективным для более широкого круга пациентов [6]. С другой стороны, задача цифрового двойника — зеркально отразить ключевые характеристики одного человека, связанные с его реакцией на конкретные ЛС. В совокупности эти вычислительные инструменты открывают широкие возможности для разработки ЛС, доз, протоколов и комбинаций, которые будут полезны для большей части пациентов [29][30]. Однако эти исследования имеют некоторые недостатки:
- ограниченный доступ к участникам исследования: могут ограничить доступ к участникам, которые не имеют доступа к необходимым технологиям или являются неудобными с использованием электронных платформ. Это может привести к искажению выборки и нерепрезентативности результатов исследования;
- ограниченный контроль над соблюдением протокола: требуют большей самостоятельности участников исследования в выполнении протокола и сборе данных. Это может привести к неправильному или неполному соблюдению протокола, что может исказить результаты исследования;
- проблемы с конфиденциальностью данных: требуют передачи и хранения большого объёма данных, включая медицинскую информацию, через электронные платформы. Это может создавать риски для конфиденциальности и безопасности данных, особенно если не принимаются соответствующие меры защиты;
- ограниченное взаимодействие с исследователями: могут ограничить возможность участников взаимодействовать с исследователями и задавать вопросы. Это может привести к недостаточному пониманию исследования и его требований, а также к потенциальным проблемам с обеспечением этической защиты и безопасности участников;
- недостаток реального контекста: могут ограничить возможность изучения воздействия ЛС и терапий в реальном медицинском окружении. Это может ограничить общую применимость результатов исследования и их способность быть перенесёнными в реальную клиническую практику.
Виртуальные испытания всё ещё являются относительно новой областью исследований, и некоторые из этих недостатков могут быть устранены или смягчены с развитием технологий и совершенствованием методологии. Быстрому развитию данного направления в США способствовали рекомендации FDA от 2021 г. [31]. Однако данный документ остаётся не утверждённым полностью, в Европе и в России аналогичные документы находятся в стадии разработки. Принятие подхода на основе ИИ может улучшить соблюдение протоколов клинических испытаний [32]. Хотя цифровизация, виртуализация и децентрализация не являются панацеей от кризисов клинических исследований, они могут повысить эффективность, что может оказать значительное и долгосрочное влияние.
ОРГАНИЗАЦИОННАЯ СТРУКТУРА В КЛИНИЧЕСКИХ ИСПЫТАНИЯХ / ORGANIZATIONAL STRUCTURE IN CLINICAL TRIALS
Основными составляющими клинических исследований являются пациенты, исследователи, академические центры, спонсоры (фармкомпании или государственные структуры), регуляторные органы, организации по защите прав пациентов и контрактно- исследовательские организации [3]. В любом случае пациент является центром вселенной клинических испытаний (рис. 3). Вся эта система нуждается в цифровом пересмотре, поскольку многие центры по-прежнему используют папки с протоколом, бумажные дневники, которые заполняются ручкой, факсимильное общение между центрами, неструктурированные данные и давние системы программного обеспечения [33]. Клинические испытания должны быть легкодоступными и должны обеспечивать такую возможность, чтобы ни один пациент не был исключён без необходимости; это может быть достигнуто с помощью агностического сопоставления исследовательских центров, клинических испытаний и навигационных услуг. Кроме того, обучение по клиническим испытаниям должно быть частью медицинского образования [3][34].

Рис. 3. Пациент как центр клинических испытаний
Fig. 3. The patient as the center of clinical trials
Примечания: Адаптировано по Subbiah V (2023) [5]: АМА — Африканское агентство по лекарственным средствам; CDSCO — центральная организация по контролю за стандартами лекарственных средств, Индия); CMS — центры Medicare и Medicaid Services; ВКГ — внешняя контрольная группа; EMA — Европейское агентство по лекарственным средствам); HTA — оценка технологий здравоохранения); NMPA — Национальное управление по лекарственным препаратам и медицинским изделиям, Китай; РЗН — Федеральная служба по надзору в сфере здравоохранения и социального развития в Российской Федерации).
Notes: Adapted by Subbiah V (2023) [5]: АМА — African Medicines Agency; CDSCO — Central Drugs Standard Control Organization, India; CMS — Centers for Medicare and Medicaid Services; ВКГ — external control group; EMA — European Medicines Agency; HTA — Health Technology Assessment; NMPA — National Medical Products Administration, China); РЗН — Federal Service for Supervision of Healthcare and Social Development in the Russian Federation.
ИЗМЕНЕНИЯ В ПОДХОДАХ РЕГУЛЯТОРОВ / CHANGES IN THE APPROACHES OF REGULATORS
Сроки клинических разработок для лекарственных средств-кандидатов в среднем составляют около 10 лет (см. рис. 1) [3]. Некоторое ускорение разработки было достигнуто на этапе молекул и рецепторных взаимодействий компьютерного моделирования, использование адаптивных дизайнов и т. д. [35]. В то же время процесс экспертизы и регуляторного одобрения по-прежнему замедляет выход ЛС на фармацевтический рынок. В то же время пандемия COVID-19 потребовала быстрой регистрации вакцин и ЛС для лечения, что привело к актуализации ускоренной разработки и регистрации новых ЛС. В Российской Федерации процедура ускоренной экспертизы и регистрации только орфанных лекарственных препаратов прописана в 26 статье Федеральном законе №61 «Об обращении лекарственных средств» от 12.04.2010 [36]. Однако пандемия COVID-19 и санкции привели к необходимости расширения списка ЛС с разрешённой ускоренной регистрацией. В России с весны 2022 г. процедура ускоренной регистрации возможна в отношении ЛС, признанных дефектурными [37][38].
Программа ускоренного одобрения ЛС была предложена в США в 1992 г. преимущественно для вывода на рынок новых методов лечения ВИЧ-инфекции до завершения испытаний. Далее FDA разработала программу ускоренного утверждения ЛС, позволяющую раньше утверждать ЛС для лечения серьёзных заболеваний и удовлетворения неудовлетворённых медицинских потребностей на основе суррогатных конечных точек. Использование суррогатной конечной точки может значительно сократить время, необходимое для получения разрешения FDA. При этом компании, производящие ЛС, всё равно обязаны провести исследования, подтверждающие предполагаемый клинический эффект. Если подтверждающие исследования показывают, что ЛС действительно приносит клиническую пользу, то FDA выдаёт традиционное разрешение на его применение. Если же подтверждающие исследования не показывают, что ЛС приносит клиническую пользу, то FDA применяет регуляторные процедуры, которые могут привести к изъятию ЛС с фармацевтического рынка [39]. За год в РФ по этой схеме был зарегистрирован 51 ЛС.
Однако ускоренная регистрация вызывает много вопросов, касающихся достаточности уровня доказательности и безопасности. По подсчётам агентства Bloomberg за прошедшие 30 лет фармкомпании подали около 300 заявок на одобрение лекарств по программе ускоренного одобрения, в основном в онкологии. В то же время FDA отозвало разрешение у семи таких ЛС. Как заявляли в Министерстве здравоохранения США, в период с 2018 по 2021 гг. федеральные программы Medicare и Medicaid потратили более $18 млрд на ЛС для ускоренной регистрации, подтверждающие испытания которых в конечном итоге были отложены. Завершение испытаний и предоставление их итогов FDA — одно из условий получения ускоренной регистрации [40].
МЕЖГОСУДАРСТВЕННАЯ ГАРМОНИЗАЦИЯ И ОБОБЩАЕМОСТЬ ДЛЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ / INTERSTATE HARMONIZATION AND GENERALIZABILITY FOR DRUGS
Существует настоятельная необходимость в глобальной гармонизации деятельности регуляторных органов разных стран для устранения существенного неравенства в доступе к ЛС. В каждом регионе мира существуют регулирующие органы, такие как FDA, Европейское агентство по лекарственным средствам (англ. European Medicines Agency; ЕМА), Федеральная служба по надзору в сфере здравоохранения и социального развития в Российской Федерации, Национальное управление по лекарственным препаратам и медицинским изделиям Китая (англ. National Medical Products Administration; NMPA), Африканское агентство по лекарственным средствам (англ. African Medicines Agency; АМА), Центральная организация по контролю за стандартами лекарственных средств Индии (англ. Central Drugs Standard Control Organization; CDSCO), Центры Medicare и Medicaid Services США (англ. Centers for Medicare and Medicaid Services; CMS) и другие. Все эти агентства работают независимо, и часто регистрация ЛС на основе исследований, проведённых спонсором из одной страны, затруднена в другой стране, где эти исследования не проводили. Однако реальность такова, что клинические исследования, включая многоцентровые РКИ, часто не могут проводиться в каждой стране для получения конкретных доказательств для населения этой страны. Анализ обобщаемости на основе специально проведённых сравнительных исследований может дать некоторые доказательства внешней валидности, влияющей на оценку местных регуляторными органами, и технологии здравоохранения [41]. На уровне стран Евразийского экономического союза (ЕАЭС) гармонизация правил обращения ЛС происходит на национальном или наднациональном уровнях путём подписания Соглашения о единых принципах и правилах обращения ЛС в рамках Евразийского экономического союза от 23.12.2014. Единые правила ЕАЭС предусматривают две процедуры регистрации лекарственных средств: процедура взаимного признания; децентрализованная процедура. В рамках процедуры взаимного признания лекарственное средство сначала регистрируется в одном из государств — членов ЕАЭС (так называемом референтном государстве) и в этом государстве получает регистрационное удостоверение. После этого лекарственное средство регистрируется в других государствах — членах ЕАЭС (так называемых государствах признания), но уже по сокращенной процедуре, так же происходит с получением регистрационного удостоверения в государстве признания. Отличие децентрализованной процедуры заключается в том, что регистрационное досье оценивается одновременно в референтном государстве и государстве признания. Это позволяет сократить общий срок регистрации в нескольких государствах [42].
ДАННЫЕ РЕАЛЬНОЙ КЛИНИЧЕСКОЙ ПРАКТИКИ И ДОКАЗАТЕЛЬСТВА, ПОЛУЧЕННЫЕ ИЗ ДАННЫХ РЕАЛЬНОЙ КЛИНИЧЕСКОЙ ПРАКТИКИ / RWD AND RWE
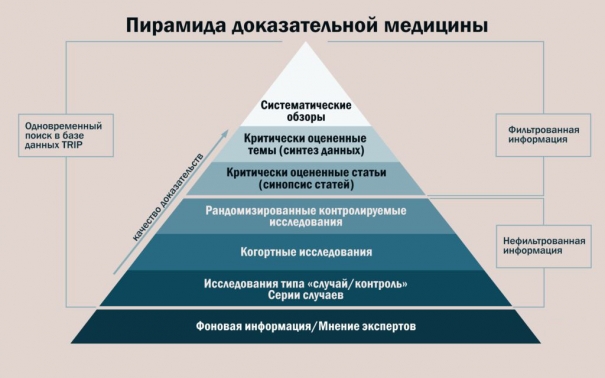
Первоначальная идея ДМ была сведена к доказательствам, предоставляемым после проведения РКИ. В то же время уже в своих первых работах Cochrane А (1972 г.) указывал, что есть существенная разница в данных РКИ и реальной пользе для пациента [43]. Действительно, сейчас хорошо известна проблема разрыва между данными предрегистрационных РКИ (действенность, efficacy) и эффектами у пациента в реальной клинической практике (эффективность, effectiveness). Более того, в 1995 году была предложена целая концепция “efficacy-effectiveness gap”, дающая объяcнение причинам таких различий [44]. Традиционные испытания были разработаны на основе ошибочного представления о том, что регуляторные органы не будут учитывать разнообразные доказательства из RWD. Важно различать RWD, которые относятся к данным, полученным при стандартном лечении пациентов, и реальные «доказательства» (RWE), которые являются доказательствами, полученными от RWD в отношении потенциального использования продукта [45][46]. К RWD можно отнести данные, получаемые после проведения больших упрощённых рандомизированных исследований, прагматических клинических исследований, пострегистрационных исследований безопасности, когортных исследований, исследований «случай-контроль», исследований с внешним контролем (внешнее управление). RWD получают из регистров, данных от страховых медицинских организаций, данных электронных медицинских карт и интегрированных электронных медицинских карт, электронных систем и баз данных, электронных платформ, данных носимых устройств, исходов сообщаемых пациентами (англ. Patient-Reported Outcomes; PRO), источники открытых данных, в том числе интернет-источники, социальные сети, включая так называемые исследования в сообществах (англ. community medical studies) (рис. 4). Регуляторные органы и экспертные группы во всём мире разрабатывают и представляют руководства для использований RWD / RWE [47]. Всё это вызвано стремительным ускорением ввода данных из нескольких потоков данных параллельно с новыми аналитическими возможностями (мультимодальный ИИ) для анализа огромного объёма данных. В России и в странах ЕАЭС аналогичные документы также разрабатываются [45][46].

Рис. 4. Научно обоснованный айсберг доказательной медицины
Fig. 4. Evidence-based iceberg of evidence-based medicine
Примечания: Адаптировано по Subbiah V (2023) [5]: РКИ — рандомизированные контролируемые испытания); ЭМК — электронные медицинские карты; PRO — исходы, сообщаемые пациентами.
Notes: Adapted by Subbiah V (2023) [5]: РКИ — randomized controlled trials; ЭМК — HER — Electronic Health Record; PRO — Patient-Reported Outcomes.
ЗАКЛЮЧЕНИЕ / CONCLUSION
В настоящее время мы являемся свидетелями перехода от «универсальной медицины», которая манипулирует в основном малыми молекулами, лекарствами-блокбастерами, которые назначаются большим количеством врачей, к «прецизионной медицине», которая в свою очередь уже широко применяет биотехнологические препараты, начинает использовать генную и клеточную терапии, а лекарства назначаются специалистами в центрах третичной помощи. Инновации и персонализированная медицина, развитие клинической фармакологии должны привести к тому, что каждая медицинская организация станет исследовательской, а все необходимые проверки качества и исследования станут частью стандарта лечения [48][49]. Система здравоохранения должна быть интегрирована в понятную систему генерации RWE, в которой клинические исследования и клиническая помощь идут рука об руку [45]. Вызывают интерес такие направления, как децентрализованные, цифровые, удалённые и «виртуальные» испытания. Методологические достижения и будущий анализ всех данных на основе ИИ могут обеспечить доказательства для того, чтобы реализовать цель персонализированной медицины — то есть, предложить правильное обращение к нужному пациенту в нужное время. Эти новые дизайны клинических исследований стремятся сделать исследования более эффективными, релевантными и применимыми к реальным условиям практики здравоохранения и пациентоориентированными [50]. Они помогают улучшить процесс разработки новых лекарств и методов лечения, а также повысить достоверность и обобщаемость результатов исследований.
Нынешняя пирамида доказательной медицины представляет собой верхушку айсберга и в лучшем случае предоставляет лишь поверхностные доказательства, касаемые той или иной медицинской технологии для пациента (рис. 4) [5]. Следовательно, необходим глубокий синтез и объединение всех имеющихся данных для достижения доказательной медицины следующего поколения. Основная задача в ближайшие два десятилетия будет заключаться в том, чтобы использовать потенциал многомерной генерации доказательств путём извлечения, сопоставления и получения больших массивов данных естественного течения заболеваний, геномики и всех других анализов омики, всех опубликованных клинических исследований, RWD для предоставления доказательств следующего поколения.
СПИСОК ЛИТЕРАТУРЫ
↑1. Вербицкая Е.В. Доказательная медицина: основные понятия, принципы поиска и оценки информации: методическое пособие / Е. В. Вербицкая; под ред. А. С. Колбина. - СПб.: РИЦ ПСПбГМУ, 2017. - 36 с. [Verbitskaya EV. Dokazatel'naya medicina: osnovnye ponyatiya, principy poiska i ocenki informacii: metodicheskoe posobie / EV Verbitskaya; ed by. AS Kolbin. SPb.: PPC PSPBGMU, 2017. (In Russ.)].
↑2. Evidence-Based Medicine Working Group. Evidence-based medicine. A new approach to teaching the practice of medicine. JAMA. 1992 Nov 4;268(17):2420-5. https://doi.org/10.1001/jama.1992.03490170092032.
↑3. Управление клиническими исследованиями / под общ. ред. Белоусова Д. Ю., Зырянова С. К., Колбина А. С. - 1-е изд. - М.: Буки Веди: Издательство ОКИ, 2017. - 676 с.: ил. - ISBN 978-5-4465-1602-5. [Upravlenie klinicheskimi issledovaniyami / Ed by Belousov DYU, Zyryanova SK, Kolbina AS. 1-e izd. Moscow: Buki Vedi: Publishing House OKI, 2017. (In Russ).].
↑4. Rethinking evidence in medicine. Nat Med. 2023 Jan;29(1):1. https://doi.org/10.1038/s41591-022-02186-3.
↑5. Subbiah V. The next generation of evidence-based medicine. Nat Med. 2023 Jan;29(1):49-58. https://doi.org/10.1038/s41591-022-02160-z.
↑6. Martin L, Hutchens M, Hawkins C, Radnov A. How much do clinical trials cost? Nat Rev Drug Discov. 2017 Jun;16(6):381-382. https://doi.org/10.1038/nrd.2017.70.
↑7. Food and Drug Administration. Adaptive Design Clinical Trials for Drugs and Biologics Guidance for Industry. 2019. [Online]. Available: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/adaptive-design-clinical-trials-drugs-and-biologics-guidance-industry.
↑8. Бондарева И. Б. Адаптивный дизайн в клинических исследованиях: преимущества и риски. Качественная клиническая практика. 2017;(3):23-34. [Bondareva IB. Adaptive design in clinical trials: benefits and risks. Kachestvennaya klinicheskaya praktika. 2017;(3):23-34. (In Russ.)]. Accessed: Jun. 19, 2023. [Online]. Available: https://www.clinvest.ru/jour/article/view/91/91.
↑9. Park JJH, Siden E, Zoratti MJ, et al. Systematic review of basket trials, umbrella trials, and platform trials: a landscape analysis of master protocols. Trials. 2019 Sep 18;20(1):572. https://doi.org/10.1186/s13063-019-3664-1.
↑10. Woodcock J, LaVange LM. Master Protocols to Study Multiple Therapies, Multiple Diseases, or Both. N Engl J Med. 2017 Jul 6;377(1):62-70. https://doi.org/10.1056/NEJMra1510062.
↑11. Dickson D, Johnson J, Bergan R, et al. The Master Observational Trial: A New Class of Master Protocol to Advance Precision Medicine. Cell. 2020 Jan 9;180(1):9-14. https://doi.org/10.1016/j.cell.2019.12.009.
↑12. Das S, Lo AW. Re-inventing drug development: A case study of the I-SPY 2 breast cancer clinical trials program. Contemp Clin Trials. 2017 Nov;62:168-174. https://doi.org/10.1016/j.cct.2017.09.002.
↑13. Redman MW, Papadimitrakopoulou VA, Minichiello K, et al. Biomarker-driven therapies for previously treated squamous non-small-cell lung cancer (Lung-MAP SWOG S1400): a biomarker-driven master protocol. Lancet Oncol. 2020 Dec;21(12):1589-1601. https://doi.org/10.1016/S1470-2045(20)30475-7.
↑14. Subbiah V, Puzanov I, Blay JY, et al. Pan-Cancer Efficacy of Vemurafenib in BRAFV600-Mutant Non-Melanoma Cancers. Cancer Discov. 2020 May;10(5):657-663. https://doi.org/10.1158/2159-8290.CD-19-1265.
↑15. Subbiah V, Kreitman RJ, Wainberg ZA, et al. Dabrafenib and Trametinib Treatment in Patients With Locally Advanced or Metastatic BRAF V600-Mutant Anaplastic Thyroid Cancer. J Clin Oncol. 2018 Jan 1;36(1):7-13. https://doi.org/10.1200/JCO.2017.73.6785.
↑16. Subbiah V, Cassier PA, Siena S, Garralda E, Paz-Ares L, Garrido P, Nadal E, Vuky J, Lopes G, Kalemkerian GP, Bowles DW, Seetharam M, Chang J, Zhang H, Green J, Zalutskaya A, Schuler M, Fan Y, Curigliano G. Pan-cancer efficacy of pralsetinib in patients with RET fusion-positive solid tumors from the phase 1/2 ARROW trial. Nat Med. 2022 Aug;28(8):1640-1645. https://doi.org/10.1038/s41591-022-01931-y.
↑17. Normand ST. The RECOVERY Platform. N Engl J Med. 2021 Feb 25; 384(8):757-758. https://doi.org/10.1056/NEJMe2025674.
↑18. Turner NC, Kingston B, Kilburn LS, et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): a multicentre, multicohort, phase 2a, platform trial. Lancet Oncol. 2020 Oct;21(10):1296-1308. https://doi.org/10.1016/S1470-2045(20)30444-7.
↑19. Gold S, Roig M, Miranda J, et al. Platform trials and the future of evaluating therapeutic behavioural interventions. Nat Rev Psychol. 2022;1(1):7-8. https://doi.org/10.1038/s44159-021-00012-0.
↑20. Dickson D, Johnson J, Bergan R, Owens R, Subbiah V, Kurzrock R. Snapshot: Trial Types in Precision Medicine. Cell. 2020 Apr 2;181(1):208-208.e1. https://doi.org/10.1016/j.cell.2020.02.032.
↑21. Guyatt G, Sackett D, Adachi J, et al. A clinician's guide for conducting randomized trials in individual patients. CMAJ. 1988 Sep 15;139(6):497-503.
↑22. Kim J, Hu C, Moufawad El Achkar C, et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N Engl J Med. 2019 Oct 24;381(17):1644-1652. https://doi.org/10.1056/NEJMoa1813279.
↑23. Ochoa D, Karim M, Ghoussaini M, et al. Human genetics evidence supports two-thirds of the 2021 FDA-approved drugs. Nat Rev Drug Discov. 2022 Aug;21(8):551. https://doi.org/10.1038/d41573-022-00120-3.
↑24. Robb MA, McInnes PM, Califf RM. Biomarkers and Surrogate Endpoints: Developing Common Terminology and Definitions. JAMA. 2016 Mar 15;315(11):1107-8. https://doi.org/10.1001/jama.2016.2240.
↑25. Landers M, Dorsey R, Saria S. Digital Endpoints: Definition, Benefits, and Current Barriers in Accelerating Development and Adoption. Digit Biomark. 2021 Sep 13;5(3):216-223. https://doi.org/10.1159/000517885.
↑26. Desai A, Subbiah V. COVID-19 Pandemic and Cancer Clinical Trial Pandemonium: Finding the Silver Lining. J Immunother Precis Oncol. 2020 Nov 16;4(2):64-66. https://doi.org/10.36401/JIPO-20-X7.
↑27. Soenksen LR, Ma Y, Zeng C, et al. Integrated multimodal artificial intelligence framework for healthcare applications. NPJ Digit Med. 2022 Sep 20;5(1):149. https://doi.org/10.1038/s41746-022-00689-4.
↑28. Holford N, Ma SC, Ploeger BA. Clinical trial simulation: a review. Clin Pharmacol Ther. 2010 Aug;88(2):166-82. https://doi.org/10.1038/clpt.2010.114.
↑29. Periodontal Market Size & Share Analysis - Industry Research Report - Growth Trends. https://www.mordorintelligence.com/industry-reports/virtual-clinical-trials-market (accessed Jul. 21, 2023).
↑30. FDA-Guidance. Digital Health Technologies for Remote Data Acquisition in Clinical Investigations, Guid. Ind. Investig. Other Stakeholders. 2021. Accessed: Jul. 21, 2023. [Online]. Available: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/digital-health-technologies-remote-data-acquisition-clinical-investigations.
↑31. Thomas KA, Kidziński Ł. Artificial intelligence can improve patients' experience in decentralized clinical trials. Nat Med. 2022 Dec;28(12):2462-2463. https://doi.org/10.1038/s41591-022-02034-4.
↑32. Wong DR, Bhattacharya S, Butte AJ. Prototype of running clinical trials in an untrustworthy environment using blockchain. Nat Commun. 2019 Feb 22;10(1):917. https://doi.org/10.1038/s41467-019-08874-y.
↑33. Семенов-Тян-Шанский В.Л., Бальцерович А.С., Сазонова А.Н., и др. 10 лет успеха - школа мониторов клинических исследований «CRA University». Ремедиум. 2021:(1);4-8. [Semenov-Tyan-Shanskiy VL, Bal’tserovich AS, Sazonova AN. CRA University - School of Clinical Trial Monitors: 10 years of success. Remedium. 2021:(1);4-8. (In Russ.)]. https://doi.org/10.21518/1561-5936-2021-1-4-8.
↑34. Романов A.Н., Кондакова О.Н., Григорьев Ф.В., и др. Вычислительные методы и компьютерные технологии в разработке лекарственных средств. Структура и динамика молекулярных систем. 2008;2(4А): 441-453. [Romanov AN, Kondakova ON, Grigoriev FV, et al. Vychislitel'nye metody i komp'yuternye tekhnologii v razrabotke lekarstvennyh sredstv. Struktura i dinamika molekulyarnyh sistem. 2008;2(4А):441-453. (In Russ.)]. [Online]. Available: https://www.researchgate.net/publication/230633896_Vycislitelnye_metody_i_komputernye_tehnologii_v_razrabotke_lekarstvennyh_sredstv.
↑35. Федеральный закон от 12.04.2010 N 61-ФЗ «Об обращении лекарственных средств» (с изменениями и дополнениями) [Federal Law No. 61-FZ of 12.04.2010 "Ob obrashchenii lekarstvennyh sredstv" (with amendments and additions) (In Russ.)]. https://base.garant.ru/12174909 (accessed Aug. 24, 2022).
↑36. Постановление Правительства РФ от 05.04.2022 N 593 «Об особенностях обращения лекарственных средств для медицинского применения в случае дефектуры или риска возникновения дефектуры лекарственных препаратов в связи с введением в отношении Российской Федерации ограничительных мер экономического характера» (с изменениями и дополнениями). [Decree of the Government of the Russian Federation of 05.04.2022 N 593 "Ob osobennostyah obrashcheniya lekarstvennyh sredstv dlya medicinskogo primeneniya v sluchae defektury ili riska vozniknoveniya defektury lekarstvennyh preparatov v svyazi s vvedeniem v otnos henii Rossijskoj Federacii ogranichitel'nyh mer ekonomicheskogo haraktera" (with amendments and additions). (In Russ.)]. https://base.garant.ru/404458632 (accessed Jul. 27, 2023).
↑37. Постановление Правительства Российской Федерации от 27.05.2023 № 824 «О внесении изменений в постановление Правительства Российской Федерации от 5 апреля 2022 г. № 593». [Resolution of the Government of the Russian Federation of 27.05.2023 No. 824 "O vnesenii izmenenij v postanovlenie Pravitel'stva Rossijskoj Federacii ot 5 aprelya 2022 g. № 593". (In Russ.)]. http://publication.pravo.gov.ru/document/0001202306050018?pageSize=100&index=1 (accessed Jul. 27, 2023).
↑38. FDA. Accelerated Approval Program FDA. 2022. https://www.fda.gov/drugs/nda-and-bla-approvals/accelerated-approval-program (accessed Jul. 27, 2023).
↑39. Langreth R, Rutherford F, Milton I, et al. Drug Companies Are Minting Billions on Unproven Treatments With FDA Shortcut. 2023. [Online]. Available: https://www.bloomberg.com/news/articles/2023-05-14/fda-fast-tracked-drugs-make-companies-billions-on-unproven-claims.
↑40. Ramagopalan SV, Popat S, Gupta A, et al. Transportability of Overall Survival Estimates From US to Canadian Patients With Advanced Non-Small Cell Lung Cancer With Implications for Regulatory and Health Technology Assessment. JAMA Netw Open. 2022 Nov 1;5(11):e2239874. https://doi.org/10.1001/ jamanetworkopen.2022.39874..
↑41. https://docs.eaeunion.org/docs/ru-ru/0127060/itia_24122014_doc.pdf
↑42. Cochrane A. (1972). Effectiveness and Efficiency: Random Reflections on Health Services. London: Nuffield Provincial Hospitals Trust.
↑43. Nordon C, Karcher H, Groenwold RH, et al. The "Efficacy-Effectiveness Gap": Historical Background and Current Conceptualization. Value Health. 2016 Jan;19(1):75-81. https://doi.org/10.1016/j.jval.2015.09.2938.
↑44. Исследования реальной клинической практики. Обновлённые рекомендации 2023 года. / под общей редакцией Колбина А.С. - М.: Издательство ОКИ, 2023. - 222 с. : ил. ISBN 978-5-907715-17-2. [Issledovaniya real'noj klinicheskoj praktiki. Obnovlyonnye rekomendacii 2023 goda. / Ed by Kolbin AS. Moscow: Publishing House OKI, 2023. (In Russ.)].
↑45. Решение Совета ЕЭК № 78 (ред. от 17.03.2022) «О внесении изменений в Правила регистрации и экспертизы лекарственных средств для медицинского применения». [Decision of the EEC Council No. 78 (ed. dated 03/17/2022) "O vnesenii izmenenij v Pravila registracii i ekspertizy lekarstvennyh sredstv dlya medicinskogo primeneniya". (In Russ.)]. Режим доступа: https://docs.eaeunion.org/docs/ru-ru/01431480/err_18032022_36.
↑46. ICH Reflection Paper. ICH Reflection paper on proposed international harmonisation of real-world evidence terminology and convergence of general principles regarding planning and reporting of studies using real-world data, with a focus on effectiveness of medicines. https://www.ich.org
↑47. Петров В.И., Толкачев Б.Е. Количественная клиническая фармакология и пациент-ориентированные технологии здравоохранения: перспективы 2030. Ведомости Научного центра экспертизы средств медицинского применения. Регуляторные исследования и экспертиза лекарственных средств. 2022;12(2):205-213. [Petrov VI, Tolkachev BE. Quantitative clinical pharmacology and patient-centered healthcare technologies: perspectives 2030. Vedomosti Nauchnogo tsentra ekspertizy sredstv meditsinskogo primeneniya. Regulyatornye issledovaniya i ekspertiza lekarstvennykh sredstv = Bulletin of the Scientific Centre for Expert Evaluation of Medicinal Products. Regulatory Research and Medicine Evaluation. 2022;12(2):205-213. (In Russ.)]. https://doi.org/10.30895/1991-2919-2022-12-2-205-213.
↑48. Сычёв Д.А. Переход к персонализированной медицине - приоритет научно-технологического развития России. Фармакогенетика и фармакогеномика. 2017;(1):3-4. [Sychev DA. Perekhod k personalizirovannoj medicine - prioritet nauchno-tekhnologicheskogo razvitiya Rossii. Farmakogenetika i farmakogenomika. 2017;(1):3-4. (In Russ.)].
↑49. Этическая экспертиза биомедицинских исследований: руководство для комитетов по этике. / под общей ред. А.Л. Хохлова. - 3-е изд., перераб. и доп. - М.: Изд-во ОКИ, 2021. - 792 с.: ил. ISBN 978-5-4465-3406-7. [Eticheskaya ekspertiza biomedicinskih issledovanij: rukovodstvo dlya komitetov po etike. / Ed by AL Khokhlov. - 3rd ed., reprint. and additional. - Moscow: Publishing House OKI, 2021. (In Russ.)].
Для цитирования:
Вербицкая Е.В., Белоусов Д.Ю., Колбин А.С. Доказательная медицина: новое в поиске доказательств. Качественная клиническая практика. 2023;(3):15-28. https://doi.org/10.37489/2588-0519-2023-3-15-28
For citation:
Verbitskaya E.V., Belousov D.Yu., Kolbin A.S. Evidence-based medicine: new in the search for evidence. Kachestvennaya Klinicheskaya Praktika = Good Clinical Practice. 2023;(3):15-28. (In Russ.) https://doi.org/10.37489/2588-0519-2023-3-15-28