.png)
Биоаналоги в лечении анемии при хронической болезни почек: потенциальная польза или неоправданный риск?

- Новости /
-
6695
Последнее десятилетие ознаменовано постоянным ростом количества биофармацевтических лекарственных средств (БЛС); по оценкам, к 2010 г. препараты, созданные на основе биотехнологий, составят около половины фармацевтических средств, получивших патенты. Первое поколение таких препаратов составляли продукты животного и растительного происхождения — бычий инсулин, стрептокиназа, стафилокиназа и др. Вслед за ними стали производить продукты человеческого происхождения, такие как гормон роста или антигемофильный фактор VIII. В последние десятилетия получены человеческие рекомбинантные продукты дезоксирибонуклеиновой кислоты (ДНК-продукты) — интерфероны γ, α и β, эритропоэтин (ЭПО), инсулин, гранулоцитостимулирующий фактор, гормон роста и ряд других. Первый человеческий белок (соматостатин), произведенный методом генной инженерии с использованием микроорганизмов (E. coli), был получен в 1977 г. Этот белок — гормон роста — предназначался для детей с его дефицитом. В 1978 г. создан рекомбинантный человеческий инсулин; в 1982 г. он стал первым биофармацевтическим препаратом, выпущенным на рынок. В 1981 г. разработан рекомбинантный интерферон γ, противовирусный препарат; в 1985–1986 гг. на рынок выведен рекомбинантный интерферон α — эффективное средство против вирусных инфекций, в частности хронических гепатитов (его также применяют в онкологии в качестве дополнительной терапии). В 1989 г. на фармацевтическом рынке появился первый препарат ЭПО для лечения ренальной анемии и анемии в онкологии, производимый в необходимых количествах овариальными клетками китайского хомячка. Таким образом, подавляющее большинство применяемых сегодня БЛС представляют собой рекомбинантные белки, полученные методом генной инженерии с помощью прокариотических клеток бактерий либо эукариотических клеток млекопитающих.
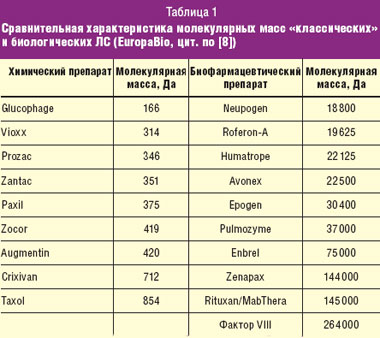
Истечение сроков патентной защиты оригинальных эпоэтинов стимулировали разработку новых версий этих продуктов, что способствует росту конкуренции и снижению цен. Окончание сроков действия патентов может стимулировать производителей инновационных продуктов на разработку и производство препаратов следующих поколений, но при этом существует опасность падения интереса к новаторству из-за возможного снижения прибыльности на фоне обостряющейся конкуренции. Появление аналогов БЛС ставит перед регуляторными органами задачу их контроля. Когда речь идет о традиционных фармпрепаратах, для вывода на рынок непатентованных дженерических лекарственных средств (ЛС) по упрощенной процедуре регистрации достаточно продемонстрировать их физико-химическое подобие и биологическую равноценность с помощью фармакокинетических и фармакодинамических исследований, проводимых на здоровых добровольцах. Такая концепция не применима по отношению к аналогам БЛС из-за трудности воспроизводства сложных белков, например таких, как ЭПО. Молекулы биологических ЛС отличаются значительно большими размерами по сравнению с веществами, полученными методом химического синтеза, и приобретают после трансляции третичную структуру. Такая молекулярная структура определяет функции этих веществ. В табл. 1 приводятся некоторые значения молекулярной массы для выборки биотехнологических препаратов в сравнении с классическими ЛС. Из таблицы видно, что в среднем молекулярная масса БЛС превосходит молекулярную массу химических препаратов в 100–1000 раз. У химических соединений конечный продукт представлен единой молекулой (или несколькими, но в очень небольшом числе), в то время как биопрепарат состоит из огромного числа смеси разных молекул. Такая смесь может быть высоковоспроизводимой при использовании мощной стандартизации и контроля на всех этапах производственного процесса, в то же время из-за сложности строения воссоздать точную копию биологического препарата практически невозможно, поскольку они не могут быть полностью «биотождественными», а только лишь «биоаналогичными». По тем же причинам невозможна адекватная характеристика биофармацевтических препаратов: невозможно адекватно охарактеризовать их с помощью существующих методов анализа.
Тем не менее в российском законодательстве до настоящего времени отсутствуют такие понятия, как «биофармацевтические» или «биологические» лекарственные средства и их «биоаналоги» [2], что не может не вызывать определенного беспокойства.
Успехи биотехнологий на примере лечения почечной анемии
Анемия при хронической болезни почек (ХБП) носит характер гипорегенераторный, нормохромный и нормоцитарный, со сниженным числом ретикулоцитов, и является полиэтиологичной патологией. Анемия особенно выражена и у пациентов на диализе: при отсутствии лечения уровень гемоглобина у 90% больных не превышает 10 г/дл. Основными причинами развития анемии при хронической почечной недостаточности (ХПН) являются недостаток выработки эндогенного ЭПО в почках, уменьшение срока жизни эритроцитов в условиях уремического окружения (гемолиз), дефицит железа. Таким образом, почечную анемию можно характеризовать как гипорегенераторную («ЭПО-дефицитную»), с признаками гемолиза и дефицита железа. Уремии свойственен как абсолютный, так и относительный дефицит эндогенного ЭПО. Концентрация ЭПО в крови больных ХПН значительно ниже, чем у больных с такой же тяжестью анемии другой этиологии. В лечении анемии широко применяются препараты рекомбинантного человеческого ЭПО (рчЭПО), которые в России используются с начала 1990-х годов [4]. Результаты рандомизированных контролируемых исследований показали, что их применение позволяет устранить анемический синдром и уменьшить потребность в гемотрансфузиях у пациентов как на преддиализной стадии, так и на стадии гемодиализа, снижает заболеваемость и смертность больных за счет сокращения сердечно-сосудистых и инфекционных осложнений, существенно повышает качество жизни и социально-трудовую реабилитацию данной категории больных [10]. Профилактика и коррекция анемии путем применения рчЭПО предотвращает и/или способствует обратному развитию гипертрофии миокарда левого желудочка, а также снижает увеличенный вследствие анемии сердечный выброс. Результаты метаанализа рандомизированных контролируемых исследований, проведенных с целью изучения эффективности применения рчЭПО у «преддиализных» и «диализных» пациентов, продемонстрировали, что использование препаратов рчЭПО способствует повышению уровня гемоглобина до рекомендуемых целевых значений (11–13 г/дл), а также снижению потребности в гемотрансфузионной терапии [3]. Применение рчЭПО, таким образом, радикально изменило не только подход к коррекции анемии почечного генеза, но и представление об адекватности заместительной терапии в целом.
Инновационные биопрепараты в лечении почечной анемии и их биоаналоги
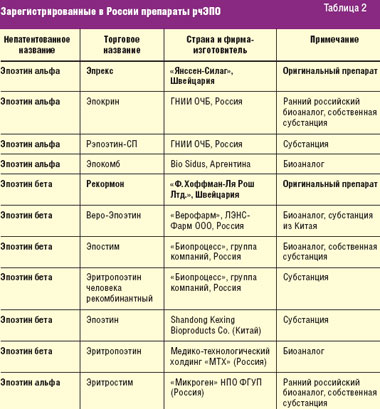
Длительное время в нашей стране использовались оригинальные БЛС — эпоэтин α и β и их первые российские аналоги, воспроизведенные еще в начале 1990-х годов. Включение в список ДЛО препаратов рчЭПО α и β, а также упрощенный порядок регистрации биоаналогов в России, не требующий обязательных клинических исследований эффективности, безопасности и иммуногенности в предрегистрационном периоде, привели к массовому выходу на российский рынок биоаналогов рчЭПО. Инновационные препараты эпоэтина α и β и их биоаналоги, зарегистрированные сегодня в РФ, представлены в табл. 2.
В ближайшие годы количество биоаналогов рчЭПО будет увеличиваться в связи с истечением срока патентной защиты оригинальных препаратов и сохраняющимся упрощенным порядком их регистрации. Наряду с отдельными весомыми преимуществами появления биоаналогов (низкая цена, доступность), особого внимания заслуживают вопросы безопасности их применения, поскольку биотехнологические препараты крайне сложно воспроизвести полностью биоэквивалентными.
Биологические, физиологические и общеклинические свойства БЛС в значительной степени зависят от процессов производства, очистки и приготовления лекарственных форм. Новаторы в области биофармацевтики приобретают опыт и накапливают данные, связанные с влиянием производственного процесса на конечный продукт, к которым у производителя биоаналога нет и не может быть доступа. Поэтому использование той же самой линии генетически однородных клеток и идентичных процессов производства, очистки и приготовления лекарственных форм не гарантируют того, что продукт будет являться биоэквивалентным исходному БЛС.
Так, ЭПО представляет собой гликопротеид с боковыми углеводными цепочками. Степень гликозилирования эпоэтина, в частности наличие свободных остатков сиаловой кислоты, изменяет его фармакокинетику и влияет на скорость выведения из организма. Кроме того, молекула ЭПО неоднородна — она состоит из смеси различных изоформ, что изначально обусловлено различиями в гликозилировании. Изоформный состав зависит от клеточных линий, условий развития культуры и процедур очистки, в результате чего возникают различные степени гликозилирования как в пределах одной клеточной линии, так и в разных линиях.
Различные изоформы ЭПО обладают разной биологической активностью [6]. Присутствие большего количества кислотных или щелочных изоформ может изменить активность ЛС. Щелочные изоформы связывают с уменьшенным периодом полувыведения.
Другая проблема, связанная с разработкой биоаналогов БЛС, — это повышение иммуногенности. Иммуногенность биофармацевтических веществ может возрастать за счет многих факторов, включая вариации последовательности технологических процессов, гликозилирования, наличия загрязняющих веществ и попадания примесей, имеют значение и приготовление лекарственных форм, хранение и погрузочно-разгрузочные работы.
Иммуногенность — это проблема особенно актуальная в случае эпоэтина, поскольку антитела к ЭПО оказывают нейтрализующее влияние на ЭПО, что приводит к прогрессирующей резистентной анемии вследствие развития истинной эритроцитарной аплазии костного мозга (PRCA). Кроме того, наличие загрязняющих веществ и попадание примесей, связанное с технологическим процессом, могут значительно изменить качество лекарства, его эффективность и профиль неблагоприятных событий.
Бразильские ученые и органы власти недавно занялись решением этой проблемы. Так, в результате проведенных проверок бразильское Управление надзора ANVISA приостановило ввоз двух препаратов рчЭПО от разных производителей [1]. Кроме того, последнее исследование, в процессе которого проводился анализ препаратов рчЭПО от разных производителей, показало различия между производственными партиями и обнаружило недопустимые уровни бактериальных эндотоксинов.
Исследования показывают, что продукты ЭПО производителей вне Европы и США сильно отличаются по составу [6–8]. Хотя это не обязательно означает, что данные продукты с клинической точки зрения уступают оригинальным препаратам, некоторые из них не соответствуют прилагаемым к ним спецификациям; такая ситуация указывает как минимум на то, что некоторые производители недостаточно качественно контролируют производственный процесс. Кроме того, содержимое ампул зачастую превышало допустимые показатели по указанной на этикетке активности [1]. Неправильная маркировка может привести к превышению дозировки и вызвать нежелательные клинические последствия.
Многие аналоги БЛС отличаются неоднородностью с точки зрения распределения изоформ. Поскольку трудно установить влияние разнообразных изоформ на общую степень активности и токсичности препарата, нормативные стандарты требуют соблюдения единообразия между различными партиями продукта. С учетом вышесказанного особую тревогу вызывает вариабельность различных партий одного и того же продукта. Несоответствие между партиями говорит о наличии крупных недостатков в производственном процессе или, что еще хуже, дает основание подразумевать разные источники того, что позиционируется как идентичный продукт.
Требования о получении разрешения на продажу биологических непатентованных препаратов производителями является предметом бурных споров, однако все чаще превалирует мнение, что подаваемые на утверждение документы должны содержать как клинические, так и биологические данные [1]. Глубина необходимых исследований, скорее всего, будет зависеть от сложности продукта, его действия, токсичности, возможного клинического эффекта, иммуногенности и возможности сравнения реакции у различных групп населения. Для того чтобы обнаружить редкие побочные явления, регистрация продукта должна быть связана с проведением пострегистрационных испытаний. Поскольку для многих биофармацевтических препаратов является обязательным постоянное хранение в охлажденных условиях на протяжении всей цепочки поставок от завода к пациенту, органы надзора и инспекции здравоохранения могут подвергнуть проверке систему дистрибуции нового поставщика.
Помимо требований к производству и дистрибуции следует учитывать, что биофармацевтические препараты, как правило, выписываются врачами, которые ожидают высокого уровня поддержки и медицинской информации со стороны компании, занимающейся продажей этих продуктов. Компании, подающие заявки на регистрацию непатентованных биологических препаратов, должны оцениваться в том числе по способности удовлетворения этих требований.
Следствием вышесказанного явилось существенное ужесточение правил регистрации биоаналогов в Европе и США: в 2006 г. EMEA (Европейское медицинское агентство) были утверждены новые требования к регистрации биоаналогов, включающие доклинические и клинические сравнительные исследования, а также дополнительный мониторинг нежелательных явлений в течение первого года применения в клинической практике [11].
В законодательстве Евросоюза понятие «биоаналог» рассматривается как продукт, не соответствующий понятию «дженерик», поскольку он представляет собой высокомолекулярный белок, и поэтому нет уверенности в его достаточном соответствии оригинальному продукту. В ЕС и США существует единое мнение о том, что ускоренная регистрация неприменима к аналогам БЛС [1]. В странах Евросоюза с 2006 г. все биоаналоги эпоэтина должны проходить полный цикл клинических испытаний перед своей регистрацией, включая фармакокинетические исследования на добровольцах, и как минимум два проспективных рандомизированных плацебо-контролируемых исследования отдельно с внутривенным и подкожным путями введения БЛС продолжительностью 6 мес и последующим анализом данных по иммуногенности в течение 12 мес [11].
Иммуногенность биоаналогов
Большинство БЛС индуцируют иммунную реакцию (иммуногенность), что, как правило, не приводит к клинически значимым последствиям. Однако в отдельных случаях последствия иммуногенности могут быть тяжелыми и даже летальными; они способны приводить не только к снижению эффективности препарата, но и вызывать аутоиммунные реакции к эндогенным молекулам.
В случае экзогенных белковых продуктов, таких как биофармацевтические продукты, экстрагированные из микробов, растений или животных, возникновение иммунной реакции на чужеродные белки вызывает образование нейтрализующих антител. Медиатором иммунного ответа выступают Т-клетки; иммунный ответ проявляется как незамедлительная реакция после первой встречи с антигеном.
При иммунном ответе на эндогенные человеческие белки (аутоантиген), например рекомбинантные ДНК-продукты, происходит образование связывающих антител. Ответ такого рода опосредуется Т-клетками через снижение иммунной толерантности, реакция возникает быстро и исчезает после прекращения терапии.
Теоретически основой иммуногенности к биофармацевтическим ЛС является их чужеродность, экзогенное происхождение либо подобие собственным антигенам. В обоих случаях клинические симптомы иммуногенности проявляются после активации секретирующих антитела В-клеток.
Существует два механизма возникновения иммуногенности. Во-первых, посторонние включения в биофармацевтическом продукте (например, эндотоксины или денатурированные белки) посылают вторичный сигнал (так называемый сигнал об опасности) Т-клеткам, которые затем посылают сигнал о начале активации В-клеткам, в результате чего нарушается иммунологическая толерантность последних.
Во-вторых, толерантность В-клеток может нарушаться вследствие независимого ответа со стороны Т-клеток. Так, если биофармацевтический продукт не обладает способностью равномерно растворяться, он может образовывать агрегаты. Иммунная система может ошибочно принять подобные агрегаты за вирусы, вследствие чего происходят активация и пролиферация В-клеток с продукцией аутореактивных связывающих антител [7].
Факторы, влияющие на иммуногенность
Факторы, связанные с препаратом. Известны связанные с продуктом и «хозяином» факторы, вызывающие иммуногенность БЛС. К ним относят следующие: структурные особенности (например, последовательность белков), наличие экзогенных или эндогенных эпитопов и степень гликозилирования, влияющая на деградацию белка, воздействие на антигенные участки и растворимость препарата. Самая высокая иммуногенность интерферона α-2 (ИФН α-2), получаемого из клеток Escherichia coli, была связана с недостаточным гликозилированием по сравнению с другими продуктами, экстрагированными из CHO-клеток (овариальных клеток китайского хомячка). Другими факторами, влияющими на иммуногенность, являются форма выпуска, условия хранения, технология производства и выделения продукта, недостаточная его очистка и наличие радионуклидных примесей. Доказательство важности указанных факторов — многочисленные зарегистрированные случаи, связанные с антигенностью препаратов ИФН α-2 от разных производителей. Было показано, что изменяя форму выпуска и условия хранения ИФН α-2, можно снизить его иммуногенность.
Факторы, связанные с «хозяином». На иммуногенность биофармацевтических продуктов влияют несколько факторов, связанных с «хозяином». Генетические особенности пациента могут приводить к образованию нейтрализующих антител. Так, аллель основного комплекса гистосовместимости влияет на способность «хозяина» распознавать антиген в реакциях, медиаторами которых являются Т-клетки. Одновременно важным фактором может оказаться генетическая последовательность с кодом эндогенного эквивалента терапевтического протеина.
В ряде работ было показано, что вероятность возникновения иммуногенности у пациентов с гемофилией А, которым назначался фактор VIII, зависит от эндогенной экспрессии белка. При разрушении фактора VIII он воспринимался как чужеродный и вызывал продукцию нейтрализующих его антител. Такая ситуация возникает значительно реже при генетических мутациях молекулы белка.
На иммуногенность могут также влиять сочетанные заболевания пациентов, особенно заболевания почек и печени. При аутоиммунных заболеваниях организм предрасположен к продукции антител к терапевтическим протеинам.
Важными факторами являются дозировка и путь введения препарата. При повышенной дозировке и более продолжительном курсе лечения повышается риск развития иммуногенности. Иммуногенность повышается в том случае, когда БЛС назначается подкожно и внутримышечно, и снижается при внутривенном и локальном назначении [7].
Последствия иммуногенности к биофармацевтическим ЛС
Во многих случаях наличие антител не приводит к каким-либо биологическим или клиническим последствиям. Но и при применении хорошо зарекомендовавших себя инновационных биофармацевтических средств известны случаи биологических и клинических последствий иммуногенности. Наиболее распространен биологический эффект потери эффективности препарата, который описан для ИФНα-2.
Утраченную эффективность можно восстановить, увеличив дозу, что и было сделано в случае лечения фактором VIII больных с гемофилией А. Клинические последствия могут проявляться как общая реакция со стороны иммунной системы — анафилаксия, аллергические реакции или сывороточная болезнь. Раньше такие реакции были достаточно распространены, но со временем стали более редкими по мере появления хорошо очищенных продуктов и внедрения более строгих правил в производстве патентованных биофармацевтических препаратов.
Наиболее серьезные осложнения возникают при нейтрализации природного белка с высокой биологической активностью. Такие последствия были описаны в случае мегакариоцитарного фактора роста (MDGF), когда нейтрализация антителами к биофармацевтическому препарату эндогенного тромбопоэтина вызвала тяжелую форму тромбоцитопении.
Резкое увеличение числа случаев истинной эритроцитарной аплазии (PRCA), вызванной антителами, за пределами США в период с 2000 по 2002 г. указывает на то, что незначительные изменения формы выпуска хорошо зарекомендовавшего себя инновационного препарата, применявшегося в течение многих лет, приводит к значительным клиническим последствиям [5]. Случаи PRCA ассоциировались с нарушением иммунной толерантности к эритропоэтину, что привело к нейтрализации образующимися антителами не только рекомбинантного белка, но и эритропоэтина пациента.
Резкое увеличение заболеваемости наблюдалось в основном при подкожном введении эпоэтина α (торговое название Eprex/Erypo) и совпало с заменой человеческого сывороточного альбумина в качестве стабилизатора на глицин и полисорбат в 1998 г. После отмены его подкожного введения заболеваемость PRCA существенно снизилась.
Для объяснения резкого роста заболеваемости PRCA был предложен ряд механизмов. По-видимому, основную роль в этом сыграла модификация формы препарата. Исследовалось также возможное влияние материала поршня шприца [5], однако полученные результаты оказались противоречивы. Это направление отрабатывалось благодаря наблюдению, что частота случаев заболевания PRCA была ниже среди пациентов, которым ЭПО вводился подкожно из шприцев с поршнями, имевшими тефлоновое покрытие. Сейчас изучается роль мицелл (полисорбат 80 + ЭПО). Вероятно, усиление иммуногенности данного продукта объясняется условиями его хранения, использования и назначения.
Без сомнения, уменьшение числа случаев PRCA к 2003 г. было связано с несколькими факторами. В ситуации с PRCA важными представляются два аспекта: качество и безопасность биофармацевтических и аналогичных биологических ЛС. Несмотря на то, что инновационный продукт использовался уже в течение многих лет, прошло немало времени, прежде чем удалось установить взаимосвязь между относительно небольшими модификациями его формы выпуска и резким увеличением числа случаев PRCA.
В отношении аналогичных биологических продуктов картина кажется еще более запутанной. Даже в том случае, когда аналогичные биологические ЛС произведены из одного и того же генетического материала, теми же методами, имеют ту же форму и упаковку, что и инновационный продукт, не может быть абсолютной уверенности в том, что они соответствуют референсному продукту. Биологический анализ вариантов ЭPO, произведенных в Индии, странах Азии и Южной Америки, показал их расхождение с исходным препаратом рчЭПО, хотя все производители заявляли о заменяемости и биоэквивалентности препаратов.
Анализ биоаналогов ЛС
Содержание. Разница в молекулярной массе аналогичных молекул может служить указанием на возможное изменение структуры и, соответственно, изменение активности продукта и другие неизвестные последствия. В то время как полученные химическим синтезом сложные вещества (до 1000 Да) могут быть оценены с точностью до 1/100 Да, молекулярная масса биофармацевтических средств (до 145000 Да) вследствие гетерогенности производственного процесса и полученных продуктов может различаться на 1000 Да. Для оценки таких свойств, как количество, молекулярная масса или распределение биологических молекул применяются такие методы анализа, как ELISAs (иммуносорбентный анализ с ферментной меткой) и хроматография. Однако эти виды анализа являются несколько ограниченными: для их проведения требуется достаточное количество (не всегда доступное) референтного материала высокого качества, а сами методы анализа зачастую не могут обеспечить полной характеристики сложных белков. Кроме того, непросто установить клиническую значимость полученных результатов. Следовательно, все выявленные различия следует интерпретировать как потенциальные факторы риска для безопасности лечения.
Биологическая активность. Даже при совпадении массы и плотности молекул аналогичного и референсного БЛС они могут существенно различаться по действию (активности и эффективности). Проверка 12 партий эпоэтина от пяти производителей показала, что их эффективность, по сравнению с указанной на этикетке, составляла от 80 до 125%. Различной эффективностью обладают даже образцы продукта, полученные от одного производителя. Наиболее надежными для сравнения биологической активности продуктов остаются методы in vivo [9].
Физико-химическая целостность. На действенность биопрепарата могут также влиять степень гликозилирования активной субстанции и возможные примеси — скопления молекул и результаты их деградации. Для установления чистоты продукта применяются разнообразные методы, в частности: SDS-PAGE (электрофорез в полиакриламидном геле), RP-HPLC (высокоэффективная жидкостная хроматография с обратной фазой), масс-спектроскопия или углеводный анализ (применяется для всех гликопротеинов). Ограниченность этих методов объясняется потребностью в достаточно больших количествах образцов, при этом их обработка и подготовка для анализа требует большого труда. Несмотря на указанные ограничивающие факторы, эти методы сыграли свою роль в установлении различий по физико-химической целостности и степени гликозилирования аналогов БЛС.
Все изложенные факты говорят о том, как трудно создать новый «биоаналог» и поддерживать его производство в соответствии с принятыми нормами.
Стабильность продукта, как правило, в первую очередь зависит от условий его хранения, которые должны быть строго определены в зависимости от характеристик продукта. Кроме того, при недостаточном контроле за процессом производства могут различаться скорость и условия распада (деградации) биофармацевтических средств от одного и того же производителя. Стабильность молекулы и ее распад изучают, помещая молекулу в среду с повышенной температурой. Если после электрофореза в геле в нестабильном продукте обнаруживаются не связанные с основной цепочкой полосы, это указывает на высокую вероятность образования соединений. Сниженная, по сравнению с референсным продуктом, стабильность ЛС может объясняться нестабильностью его формы или активной субстанции.
Форма выпуска биофармацевтического средства не менее важна, чем его активный ингредиент. Как правило, для стабилизации белковых препаратов используются добавки, например человеческий сывороточный альбумин, эфиры полиоксиэтиленовой жирной кислоты (полисорбат), глицин, аминокислоты, хлорид кальция и мочевина. Однако добавки способны влиять на профиль безопасности препарата. Результаты начального анализа активной субстанции аналога БЛС могут указывать на допустимое сходство с референсным БЛС, но в окончательной форме выпуска могут быть установлены факторы риска. По спектрам поглощения различных форм аналогичных БЛС можно выявить различия между ними и идентифицировать химические вещества и примеси, которые могли попасть в окончательную форму. Именно такие посторонние включения или форма включающих эпоэтин мицелл с высоким содержанием полисорбата 80, а также другие обстоятельства послужили причиной увеличения числа случаев истинной эритроцитарной аплазии (PRCA) после изменения формы препарата Eprex [5].
Анализ иммуногенных реакций. Наиболее важным вопросом по безопасности аналогичных биологических продуктов является их иммуногенность. До официальной регистрации должны быть проведены сравнительные исследования различных партий продуктов между собой и с разрешенным референсным БЛС. При этом чувствительность и специфичность анализов, применяемых для проверки иммунного ответа, могут оказаться недостаточными для того, чтобы предсказать редкие случаи иммуногенной реакции. Кроме того, при отсутствии стандартизованных и валидированных методов и способов представления данных задача сравнения титров антител из разных лабораторий становится трудновыполнимой.
Традиционные экспериментальные модели могут оказаться полезными для изучения иммуногенности различных форм человеческих рекомбинантных белков, тогда как после применения методов генной инженерии, с помощью трансгенных моделей на животных, обладающих иммунной толерантностью к человеческим белкам, можно было бы получать информацию для изучения нарушений иммунной толерантности. Пока стандартизованные «животные» модели недоступны. По этой причине их предсказательная сила относительно иммуногенности продуктов является все еще достаточно ограниченной, а задача сравнения результатов, полученных в разных лабораториях, практически невыполнима. Подобные модели могут применяться во время разработки продукта, с их помощью могут определяться условия хранения и использования аналогичных биологических ЛС.
Радиоиммунопреципитация (RIPA) — это чувствительный способ тестирования, позволяющий определить высокое аффинное число антител. Однако необходимость использования радиоактивных материалов и создания трудоемких протоколов делает этот способ неподходящим для крупномасштабного скрининга. Кроме того, с помощью этого метода невозможно распознать все изотипы антител с равной чувствительностью — в основном выявляются антитела класса иммуноглобулина G.
Мнения о целесообразности применения метода ELISA для тестирования сыворотки на иммуногенность расходятся. Недавно для скрининга новых продуктов был утвержден двойной антигенный метод ELISA. Данный тест является эффективным и чувствительным методом и способен распознавать все изотипы антител. Кроме того, он хорошо коррелирует с RIPA. Поскольку само по себе наличие антител не указывает на отрицательные клинические исходы, необходимо также установить их нейтрализующее действие.
Недавно валидирован метод биопроб ЭПО, показавший его умеренную чувствительность. Как и RIPA, при высоких концентрациях ЭПО этот анализ дает ложноотрицательные результаты; по данной причине для его проведения требуется достаточный объем тестируемых образцов сыворотки. Для получения неклинических характеристик аналогов БЛС необходимо пользоваться наиболее совершенными из имеющихся методов. В силу своей ограниченности указанные тесты могут лишь дополнять клинические исследования, но не должны их замещать. При проведении аналитических тестов или клинических исследований до официальной регистрации препарата иногда не удается предсказать редкие случаи тяжелых нежелательных явлений с продолжительным инкубационным периодом. Потому после выдачи такого разрешения должен осуществляться фармконтроль, что поможет снизить риск нежелательных явлений, таких как иммуногенность [9].
Фармконтроль. Фармконтроль предполагает обнаружение, оценку, изучение и предотвращение нежелательных явлений после появления продукта на рынке. Тщательный фармконтроль за препаратом после получения разрешения на его маркетинг является добровольным мероприятием; однако осуществление фармконтроля служит интересам производителя аналогов БЛС, так как обеспечивает долгосрочные гарантии относительно его качества, безопасности и эффективности. Согласно руководству EMEA, разработанный план фармконтроля должен быть представлен в регуляторные органы вместе с пакетом данных во время утверждения биоаналога [11]. Применение фармконтроля, тем не менее, не позволило быстро среагировать на увеличение числа случаев PRCA. Это ставит под сомнение пригодность существующей системы для продолжительной постмаркетинговой оценки аналогичных биологических препаратов. Кроме того, для установления связи между возникновением этого редкого заболевания и применяемым способом лечения должно быть достаточно всего нескольких случаев. Предсказательные модели, которые применяются в подобных ситуациях, весьма различаются по точности. Чтобы в дальнейшем использовать эти модели как критерии для принятия решения, их необходимо усовершенствовать.
Основой для официальной регистрации может являться подробный профиль препарата-биоаналога. Данные по безопасности должны дополняться детальными протоколами из баз данных клиник и результатами регулярных тестов. Необходимо разработать целостную программу с допуском риска, компонентами которой будут проведение сравнительных исследований биоаналога до и после официальной регистрации и тщательный фармконтроль. Для обеспечения тождественности различных партий продукта должен осуществляться тщательный мониторинг за процессом производства. В случае обнаружения отличий в произведенном биоаналоге могут потребоваться дополнительные исследования, в том числе обеспечивающие клинические доказательства неизменности его профиля безопасности и эффективности. Важно также определить, кто, когда, что и как должен делать. Такая стратегия должна, в частности, применяться в той части программы, где речь идет о возможном риске: необходимы гарантии того, что в случае увеличения числа заболеваний среди пациентов и подозрении на связь ситуации с применением биоаналога реакция регуляторных органов последует незамедлительно [9, 11].
Выводы
Грядущее массовое появление на рынке аналогичных БЛС ведет к изменениям всей индустрии биотехнологии и медицины. Накопленный в отношении патентованных БЛС опыт диктует необходимость усиления контроля над процессами производства рекомбинантных белков. Такие белки имеют значительно более сложные молекулы, чем традиционные препараты, полученные химическим синтезом. Производство рекомбинантных белков предполагает чрезвычайно сложные процессы, в конечном итоге определяющие свойства изготовленного ЛС. Различия в процессах производства неизменно приводят к различиям биопрепаратов, которые могут обладать разным профилем клинической эффективности и безопасности; при этом такие различия не всегда удается охарактеризовать с помощью доступных аналитических методов. Процесс производства аналогов БЛС должен подвергаться всестороннему контролю.
Для надежного тестирования биоаналогов в будущем необходимы стандартизация и валидация всех видов анализа и способов представления данных. Однако всесторонние данные, свидетельствующие о качестве биоаналога, могут быть получены только в результате пререгистрационных клинических исследований и осуществления плана по фармконтролю после регистрации ЛС.
Таким образом, сегодня назрела насущная необходимость внесения изменений в действующее законодательство. В первоочередном порядке следует ввести понятия «биофармацевтические лекарственные средства (БЛС)» и «биоаналоги» (аналоги БЛС) в Федеральный закон «О лекарственных средствах». Необходимо, чтобы регуляторные и надзорные органы РФ еще до официальной регистрации препарата требовали от производителей представить результаты как минимум двух клинических исследований, подтверждающих эффективность и безопасность аналогов БЛС. После регистрации биоаналогов, как и любых БЛС, целесообразно осуществлять длительный мониторинг неблагоприятных явлений и проводить экспертную оценку их возможной связи с применяемым ЛС.
Литература
- Белоусов Д.Ю., Центр фармакоэкономических исследований, г. Москва, Качественная клиническая практика, №2, 2006 г., стр. 80-83
- Федеральный закон от 22 июня 1998 г. № 86-ФЗ «О лекарственных средствах», с изменениями, внесенными: Федеральным законом от 2 января 2000 г. № 5-ФЗ (Российская газета. № 4. 06.01.2000); Федеральным законом от 30 декабря 2001 г. № 196-ФЗ (Российская газета. № 256, 31.12.2001); Федеральным законом от 10 января 2003 г. № 15-ФЗ (Российская газета. № 5, 15.01.2003); Федеральным законом от 30 июня 2003 г. № 86-ФЗ (Российская газета. № 126, 01.07.2003) (вступил в силу с 1 июля 2003 г.); Федеральным законом от 22 августа 2004 г. № 122-ФЗ (Российская газета. № 188, 1.08.2004); Федеральным законом от 16 октября 2006 г. № 160-ФЗ (Российская газета. № 233, 18.10.2006).
- Пересмотренные Европейские рекомендации по оптимальной практике лечения анемии у пациентов с хронической почечной недостаточностью. REBPG for the Management of Anemia in Patients with Chronic Renal Failure (пер. с англ.) // Анемия, 2005. № 3. С. 1–60.
- Российские национальные рекомендации по диагностике и лечению анемии при хронической болезни почек // Анемия, 2006. № 3. С. 3–19.
- Boven K., Knight J., Bader F. et al. Epoetin-associated pure red cell aplasia in patients with chronic kidney disease: solving the mystery // Nephrol. Dial. Transplant., 2005; 20(3): 33–40.
- Jelkmann W. Recombinant EPO production–points the nephrologist should know // Nephrology. Dialysis. Transplantation. 2007; 22(10): 2749–2753.
- Kessler M., Goldsmith D., Schellekens H. Immunogenicity of biopharmaceuticals // Nephrology. Dialysis. Transplantation. 2006; 21(5): 9–12.
- Kuhlmann M., Covic A. The protein science of biosimilars // Nephrology. Dialysis. Transplantation. 2006; 21(5): 4–8.
- Locatelli1 F., Roger S. Comparative testing and pharmacovigilance of biosimilars // Nephrology. Dialysis. Transplantation. 2006; 21(5): 13–16.
- NKF-KDOQI Clinical Practice Guidelines for anemia of chronic kidney disease: update 2000. New-York, 2001, by the National Kidney Foundation, Inc.
- Wiecek A., Ashraf M. European regulatory guidelines for biosimilars // Nephrology. Dialysis. Transplantation. 2006; 21(5): 17–20.
В. Ю. Шило, кандидат медицинских наук
Центр диализа при ГКБ № 20, Москва