.png)
Изучение биоэквивалентности лекарственных средств как одного из видов клинических испытаний

- Библиотека /
-
4003
В настоящее время в Украине значительно возросло количество заявок на регистрацию отечественных и зарубежных лекарственных препаратов. Подавляющее большинство из них — генерические препараты, т.е. лекарственные средства, выпускаемые различными фармацевтическими компаниями после прекращения срока действия патента на оригинальный препарат.
Клиническая практика показала, что препараты, имеющие одни и те же активные вещества и даже лекарственные формы и дозы, но выпускаемые различными производителями, существенно различаются как по терапевтической эффективности, так и по частоте и выраженности вызываемых ими побочных эффектов (Белоусов Ю.Б., Моисеев В.С., Лепахин В.К., 1997).
В последние годы в результате развития фармакологии, внедрения высокочувствительных методов исследования, таких, как жидкостная и газовая хроматография, радиоиммунный и ферментно-химический анализ, стало возможным в полной мере понять и оценить роль особенностей технологии производства, качественного и количественного состава вспомогательных веществ лекарственной формы и многих других факторов в действии лекарственных средств.
В большинстве случаев различия в терапевтической эффективности препаратов, содержащих одни и те же активные вещества, обусловлены изменением их биодоступности — количества лекарственного вещества, которое попадает в системный кровоток, и скорости, с которой этот процесс происходит. В связи с этим возникло новое понятие — биоэквивалентность.
Два лекарственных препарата считают биоэквивалентными, если они фармацевтически эквивалентны, имеют одинаковую биодоступность и после назначения в одинаковой дозе являются сходными, обеспечивая должную эффективность и безопасность (WHO, 1994, 1996).
Оценку биоэквивалентности лекарственных средств в настоящее время считают одним из основных методов медико-биологического контроля качества генерических препаратов, т.е. таких лекарственных средств, которые содержат одно и то же активное вещество в одинаковой дозе и в той же лекарственной форме, что и соответствующее оригинальное средство.
Фармакотерапевтическое действие препарата зависит от многих факторов (биодоступность, состояние печени и почек и т.п.). Особенно трудно бывает предсказать возможную концентрацию лекарственного средства в крови при:
· наличии сопутствующих заболеваний;
· применении взаимодействующих препаратов;
· нарушении всасывания и низкой биодоступности;
· заболевании печени и почек;
· нарушении связывания лекарственных веществ с белками крови;
· генетически обусловленных особенностях метаболизма препаратов.
В таких ситуациях концентрация препарата может оказаться слишком низкой или слишком высокой. В первом случае снижается эффективность лечения, во втором — повышается риск возникновения побочных реакций.
В связи с этим при исследовании биоэквивалентности необходимо очень тщательно подходить к подбору его участников.
Прежде всего необходимо подчеркнуть, что изучение биоэквивалентности — это клинические испытания, где субъектом исследования является человек. Поэтому к таким исследованиям предъявляют те же требования, что и ко всем другим клиническим испытаниям.
Изучение биоэквивалентности следует проводить в соответствии с принципами Надлежащей клинической практики в целях гарантии качества представляемых данных и защиты прав, здоровья и благополучия исследуемых.
ВОЗ рекомендует следующие подходы к отбору испытуемых (1994).
Контингент исследуемых для изучения биоэквивалентности должен быть максимально однородным. Чтобы снизить разброс получаемых данных, в основном это должны быть здоровые добровольцы. Должны быть представлены точные критерии включения/исключения. Желательно, чтобы в исследовании участвовали мужчины и женщины (для женщин оценку риска нужно определять индивидуально и они должны быть предупреждены о возможном риске для плода в случае беременности). В исследование включают лиц в возрасте от 18 до 55 лет. Масса их тела должна быть в пределах возрастной нормы для данного пола. Предпочтительно, чтобы испытуемые были некурящими. В противном случае они должны быть идентифицированы как таковые. Пригодность добровольцев к участию в исследовании должна быть подтверждена с помощью стандартных лабораторных тестов, данных анамнеза и физического обследования. До и в процессе исследования можно проводить специальные медицинские обследования, необходимость которых обусловлена особенностями фармакологических свойств изучаемого препарата.
В некоторых случаях корректнее включать в исследование пациентов, а не здоровых добровольцев. Такая ситуация может возникнуть, если исследуемое лекарственное средство обладает известными побочными действиями и здоровью добровольцев может быть нанесен серьезный ущерб (например, при изучении лекарственных препаратов, используемых в онкологии, при лечении ВИЧ-инфекции).
При исследовании биоэквивалентности минимальное число испытуемых — 12 человек.
Проведение исследования и его дизайн должны базироваться на знаниях фармакокинетики и фармакодинамики исследуемого лекарственного вещества.
При этом должны быть обеспечены стандартные условия для испытуемых на период проведения исследования, а именно:
· пищевой и водный режим (стандартная диета);
· полное исключение или ограничение приема лекарственных средств до и в период проведения исследования;
· двигательный режим;
· режим дня;
· исключение употребления алкоголя, кофеина, наркотических средств, концентрированных соков;
· время пребывания в исследовательском центре;
· время окончания исследования.
Особенностью дизайна таких исследований является то, что каждый испытуемый получает как стандартный препарат, так и тестируемый. Отдают предпочтение перекрестному методу с рандомизированным распределением добровольцев.
При изучении биоэквивалентности лекарственных препаратов наиболее важными являются следующие фармакокинетические параметры (рис.1):
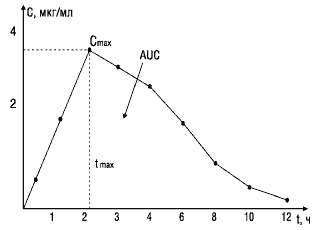
Рис.1. Основные параметры фармакокинетики, используемые при изучении биоэквивалентности лекарственных веществ
Cmax — максимум, или пик концентрации лекарственного вещества в крови;
tmax — время достижения максимальной концентрации вещества в плазме крови;
AUC — площадь под фармакокинетической кривой — кривой «концентрация—время» (изменение концентрации активного вещества в плазме или сыворотке крови во времени).
Значение показателя максимальной концентрации вещества можно объяснить с помощью следующего примера. На рис. 2 представлены фармакокинетические кривые двух лекарственных препаратов. Кривая 1 характеризует концентрацию в крови стандартного препарата, кривая 2 — тестируемого. Горизонтальной линией отмечена минимальная эффективная концентрация, при которой данное вещество оказывает терапевтическое действие. Как видно, Сmax тестируемого препарата (кривая 2) не достигает уровня минимальной эффективной концентрации и, следовательно, не оказывает терапевтического действия.
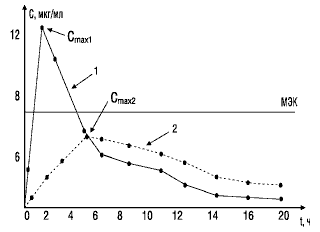
Рис. 2. Сопоставление максимальных концентраций в крови двух препаратов: МЭК — минимальная эффективная концентрация; 1 — стандартный препарат; 2 — тестируемый препарат; Сmax1, Сmax2 — соответствующие максимальные концентрации сравниваемых препаратов в крови
Второй важный параметр — время достижения максимальной концентрации лекарственного вещества в крови. Этот показатель отражает скорость его всасывания и скорость наступления терапевтического эффекта. На рис. 3 показано, что Сmax стандартного препарата (кривая 1) достигается через 1 ч, а тестируемого (кривая 2) — через 4 ч. Такое различие во времени достижения максимальной концентрации препарата в крови может обусловить изменение клинических показаний к применению данного препарата.
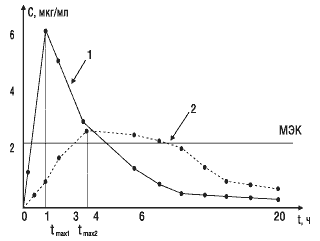
Рис.3. Сопоставление времени достижения максимальной концентрации двух препаратов: МЭК — минимальная эффективная концентрация; 1 — стандартный препарат; 2 — тестируемый препарат; tmax1, tmax2 — соответствующее время достижения максимальных концентраций сравниваемых препаратов в крови
Третьим важным параметром биоэквивалентности является площадь под фармакокинетической кривой, которая отражает количество вещества, поступившего в кровь после однократного введения препарата. На рис. 3 показано, что две кривые имеют разную форму, разные пики и неодинаковое время достижения максимальной концентрации. Но площади под этими кривыми близки по величине, следовательно, оба препарата обеспечивают поступление в кровь одинакового количества лекарственного вещества.
На рис. 4 представлен другой пример соотношения кривых, отражающих кинетику двух сравниваемых препаратов. Площадь под кривой 1 практически в два раза больше, чем под кривой 2. Обращает на себя внимание то, что максимальная концентрация и время ее достижения схожи у стандартного и тестируемого препаратов. Однако площадь под фармакокинетической кривой у тестируемого препарата в 2 раза меньше за счет более быстрого выведения его из крови. В данном случае можно ожидать уменьшения длительности действия лекарственного препарата и снижения его терапевтического эффекта.
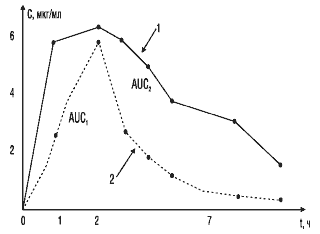
Рис. 4. Сопоставление площадей под фармакокинетическими кривыми двух препаратов: 1 — стандартный препарат; 2 — тестируемый препарат; AUC1, AUC2 — соответствующие площади под кривыми «концентрация—время» сравниваемых препаратов
Таким образом, два препарата считают биоэквивалентными, если они имеют схожие фармакокинетические показатели. По регламенту ВОЗ (1994, 1996) и ЕС (1992) их различие не должно превышать 20%.
Необходимо отметить, что планировать и проводить исследование по определению биоэквивалентности должен коллектив специалистов различных профилей: клинические фармакологи, врачи-клиницисты, биохимики, химики-аналитики, биостатистики. Все этапы проведения исследования должны быть тщательно описаны, проанализированы и представлены в подробном отчете.
Внедрение определения биоэквивалентности как метода позволяет сделать обоснованное заключение о качестве, эффективности и безопасности сравниваемых препаратов на основании меньшего объема первичной информации и в более сжатые сроки, чем при проведении клинических испытаний.
В настоящее время уже существуют регламенты изучения биоэквивалентности ВОЗ (1996), ЕС (1992), Российской Федерации (1995). В них изложены основные обоснования необходимости проведения таких исследований.
Так, изучение биоэквивалентности проводят, если:
· существует риск отсутствия биоэквивалентности и/или
· существует риск снижения фармакотерапевтического действия и клинической безопасности препарата.
Например, обязательно оценивают:
· препараты для лечения состояний, при которых необходим гарантированный терапевтический эффект;
· препараты с узким терапевтическим коридором безопасности;
· препараты, фармакокинетика которых осложнена снижением абсорбции менее 70% или с высокой элиминацией (более 70%);
· препараты с неудовлетворительными физико-химическими свойствами (низкая растворимость, нестабильность, полиморфизм);
· препараты с документированно подтвержденным существованием проблем биодоступности.
В настоящее время в Украине достаточно широко используют высокоэффективные методы для определения фармакокинетических параметров. Расширение имеющейся материально-технической базы, разработка регламента проведения исследований биоэквивалентности лекарственных веществ, подготовка специалистов в этой области позволят решить актуальную задачу по оценке эффективности и безопасности генерических препаратов отечественного и зарубежного производства.
В.И. Мальцев, А.П. Викторов,
В.Н. Коваленко, Т.К. Ефимцева, Л.И. Ковтун
Государственный фармакологический центр Министерства здравоохранения Украины, Киев
ЛИТЕРАТУРА | |
|
Источник: apteka.ua